Аутосомно рецессивный тип наследования примеры болезней

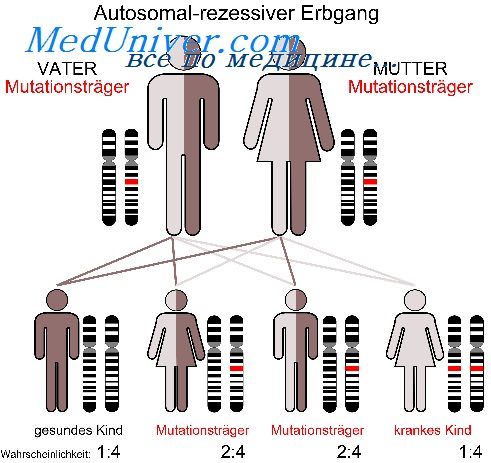
Чаще всего аутосомно-рецессивные заболевания проявляют себя при браке типа Аа х Аа (оба родителя фенотипически здоровы, но являются носителями мутантного гена) в варианте образования гомозиготного организма (25%) по дефектному гену (рис.)
Пример родословной с аутосомно-рецессивным типом наследования заболеваниямуковисцидоз– рис.
По аутосомно-рецессивному типу наследуется абсолютное большинство ферментопатий (наследственных заболеваний обмена веществ). Наиболее частыми и значимыми в клиническом отношении являются такие болезни с аутосомно-рецессивным типом наследования, как муковисцидоз (кистофиброз поджелудочной железы), фенилкетонурия, адреногенитальный синдром, многие формы нарушения слуха или зрения, болезни накопления.
Муковисцидоз(кистофиброз поджелудочной железы). Ген муковисцидоза, контролирующий синтез белка (трансмембранного регулятора проводимости) расположен на 7-й хромосоме. Заболевание обусловлено генерализованным поражением экзокринных желез. При отсутствии синтеза трансмембранного регулятора (первичного продукта гена) нарушается транспорт хлоридов в эпителиальных клетках, что приводит к избыточному выведению хлоридов, следствием чего является гиперсекреция густой слизи в клетках поджелудочной железы, бронхов, слизистой оболочке желудочно-кишечного тракта. Выводные протоки поджелудочной железы закупориваются, слизь не выводится, образуются кисты. Ферменты поджелудочной железы не поступают в просвет кишечника. Гиперпродукция слизи в бронхиальном дереве ведет к закупорке мелких бронхов и последующему присоединению инфекции. Подобные процессы развиваются в придаточных пазухах носа и в канальцах семенников. В потовой жидкости повышена концентрация ионов натрия и хлора, что и является основным диагностическим лабораторным тестом. Интеллектуальное развитие у детей не страдает. Прогноз жизни при всех формах заболевания неблагоприятный. В настоящее время пациенты со смешанными формами редко живут более 20 лет. Частота его среди новорожденных в европейской популяции составляет 1:2500.
Среди заболеваний с аутосомно-рецессивным типом наследования встречаются те, которые поражают различные системы органов и связаны с наличием комбинированных дефектов. Например, при синдроме Барде-Бидля наблюдается сочетанное нарушение умственного развития и зрения.Признаками синдрома являются: ожирение, гипогенитализм (нередко и гипогонидизм), умственная отсталость, пигментная дегенерация сетчатки, приводящая к потере зрения, и полидикталия со стороны 5 пальца. Из других нарушений зрения описаны катаракта, атрофия зрительных нервов, нистагм. Уже на первом году жизни развивается ожирение, которое с возрастом прогрессирует. Характерным для синдрома является разнообразная патология почек.
Вероятность проявления таких заболеваний кратно возрастает при кровнородственных браках. (рис. ).
В целом, в характеристике аутосомно-рецессивных заболеваний присутствуют следующие составляющие:
1) родители больного ребенка фенотипически здоровы, но являются носителями аномального гена в рецессивном состоянии;
2) мальчики и девочки заболевают одинаково часто;
3) риск рождения ребенка с аутосомно-рецессивным заболеванием составляет 25%;
4) отмечается «горизонтальное» распределение больных в родословной, т. е. пациенты чаще встречаются в рамках одного сибства;
5) наблюдается увеличение частоты больных детей в группах родителей, связанных родством, причем чем реже аутосомно-рецессивное заболевание встречается в популяции, тем чаще больные происходят из кровнородственных браков.
Источник
Аутосомно-рецессивное наследование. Пример аутосомно-рецессивного наследованияВ классических случаях аутосомно-рецессивного наследования генотип родителей больных имеет вид Аа х Аа (где а — рецессивный мутантный ген А — доминантный нормальный ген). Такой тип наследования характерен для болезни Фридрейха, гепатолентикулярной дегенерации, спинальной амиотрофии Верднига-Гофманаи Кугельберга-Веландер, атаксии-телеангиэктазии и ряда других моногенных заболеваний нервной системы. При проведении генеалогического анализа в семьях с предполагаемым аутосомно-рецессивным типом наследования следует принимать во внимание одно важное обстоятельство. Как было указано выше, в соответствии с менделевскими законами доля сибсов, пораженных аутосомно-рецессивным заболеванием, должна составлять около 1/4 от общего числа детей в поколении. Поскольку для современной структуры семей характерно сравнительно небольшое число детей (1-3 ребенка), в большинстве случаев аутосомно-рецессивные болезни проявляются в виде единичных (снорадических) случаев, и наследственно-семейный характер заболевания бывает очевидным далеко не всегда. В такой ситуации отсутствие семейного анамнеза не снимает вопрос о генетической природе болезни и не исключает 25%-й риск появления ее повторных случаев при рождении других детей у данной родительской пары.
Еще один источник ошибок в оценке данного типа наследования проиллюстрирован на рис. 10 на примере большой семьи с аутосомно-рецессивнои мышечной дистрофией, обследованной нами в одном из горных изолятов Северного Кавказа. В данной высокоинбредной семье заболевание наблюдается у 12 родственников из 3 разных поколений, что, на первый взгляд, противоречит модели аутосомно-рецессивного наследования. ()днако ни в одном случае в данной родословной нет прямой передачи болезни от родителей детям, а в пределах каждой конкретной родительской пары характер сегрегации подчиняется всем закономерностям, характерным для аутосомно-рецессивной патологии. Таким образом, сам по себе факт наличия болезни в нескольких поколениях обширной родословной не исключает аутосомно-рецессивный тип наследования, и ключевым при-знаком здесь является манифестация симптомов у части потомства (-25%) при клинически здоровых родителях, являющихся облигатными гетерозиготными носителями мутации. Правило об отсутствии прямой передачи аутосомно-рецессивной болезни в следующее поколение имеет редкие исключения: такое бывает возможным в ситуации, когда больной вступает в брак либо с другим больным с тем же заболеванием (тип брака аа х аа), либо с гетерозиготным носителем мутации в том же гене (аа х аА). В первом случае все дети унаследуют 2 копии мутантного гена и будут больными, во втором случае заболеет половина детей. Пример представлен на рисунке (показана родословная наблюдавшейся нами семьи, отягощенной болезнью Фридрейха). — Также рекомендуем «Х-сцепленное рецессивное наследование. Пример X-сцепленного наследования» Оглавление темы «Наследование неврологических заболеваний»: |
Источник
Чаще всего патологии передает тип наследования аутосомно-доминантный. Это моногенное наследование одного из признаков. Помимо этого, болезни могут передаваться детям аутосомно-рецессивным и аутосомно-доминантным типом наследования, а также по митохондриальному признаку.

Типы наследования
Моногенное наследование гена может быть рецессивным и доминантным, митохондриальным, аутосомным или сцепленным с половыми хромосомами. При скрещивании может получиться потомство с самыми разными типами признаков:
- аутосомно-рецессивными;
- аутосомно-доминантными;
- митохондриальными;
- Х-доминантное сцепление;
- Х-рецессивное сцепление;
- У-сцепление.
Разные типы наследования признаков — аутосомно-доминантный, аутосомно-рецессивный и другие, способны передавать разным поколениям мутантные гены.
Особенности наследования аутосомно-доминантного типа
Аутосомно-доминантный тип наследования заболевания характеризуется передачей мутантного гена в гетерозиготном состоянии. У потомства, получившего мутантный аллель, может проявиться генное заболевание. При этом вероятность проявления измененного гена у мужчин и женщин одинаковая.
При проявлении у гетерозигот признак наследования не оказывает серьезного влияния на здоровье и функцию к воспроизводству. Гомозиготы с мутантным генном, который передал тип наследования аутосомно-доминантный, как правило, нежизнеспособны.
У родителей мутантный ген располагается в половой гамете совместно со здоровыми клетками, и вероятность его получения у детей будет равняться 50 %. Если доминантный аллель будет изменен не полностью, то дети таких родителей будут полностью здоровы на генном уровне. При низком уровне пенентрантности мутантный ген может проявляться не в каждом поколении.
Чаще всего тип наследования аутосомно-доминантный передает заболевания из поколения в поколение. При этом виде наследования у больного ребенка один из родителей болеет тем же заболеванием. Однако, если в семье болеет только один родитель, а второй имеет здоровые гены, то дети могут и не получить по наследству мутантный ген.
Пример наследования по аутосомно-доминантному типу
Тип наследования аутосомно-доминантный может передавать более 500 разных патологий, среди них: синдром Марфана, Элерса-Данло, дистрофия, болезнь Реклингхайзена, Гентингтона.
При изучении родословной можно проследить аутосомно-доминантный тип наследования. Примеры этому могут быть разные, но самый яркий – это болезнь Гентингтона. Она характеризуется патологическими изменениями нервных клеток в структурах переднего мозга. Проявляется недуг забывчивостью, слабоумием, проявлением непроизвольных телодвижений. Чаще всего это заболевание проявляется после 50 лет.

При прослеживании родословной можно выяснить, что хотя бы один из родителей страдал такой же патологией и передал ее по аутосомно-доминантному типу. Если же у больного есть брат или сестра единокровные, но у них нет проявления болезни, значит, родители передали патологию по гетерозиготному признаку Аа, при котором генные нарушения встречаются у 50 % детей. Следовательно, у потомства больного также могут родиться 50 % детей с видоизмененным геном Аа.
Аутосомно-рецессивный тип
При аутосомно-рецессивном наследовании отец и мать являются носителями патогена. У таких родителей 50 % детей рождаются носителями, 25 % — здоровыми и столько же — больными. Вероятность передачи патологического признака девочкам и мальчикам одинакова. Однако заболевания аутосомно-рецессивного характера могут передаться не каждому поколению, а проявляться через одно-два поколения потомства.
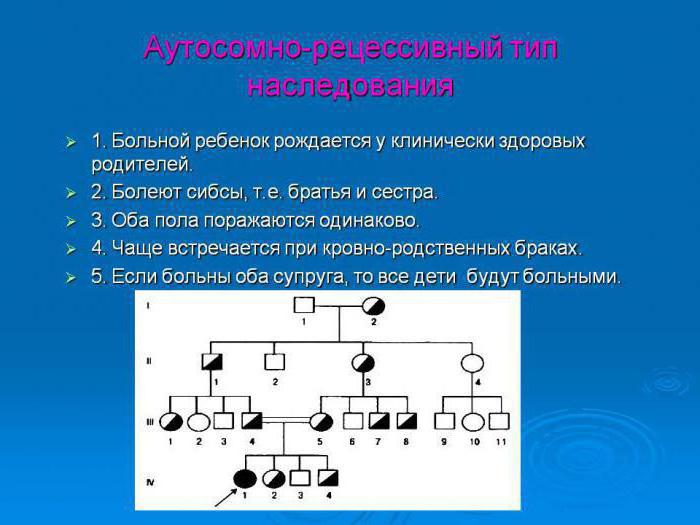
Примером заболеваний, передающихся по аутосомно-рецессивному типу, могут быть:
- болезнь Тоя-Сакса;
- нарушения обмена веществ;
- муковисцидоз и пр.
При обнаружении детей с аутосомно-рецессивным типом генных патологий выясняется, что родители находятся в родственной связи. Такое часто наблюдается в закрытых поселениях, а также в местах, где разрешаются кровные браки.
Х-хромосомное наследование
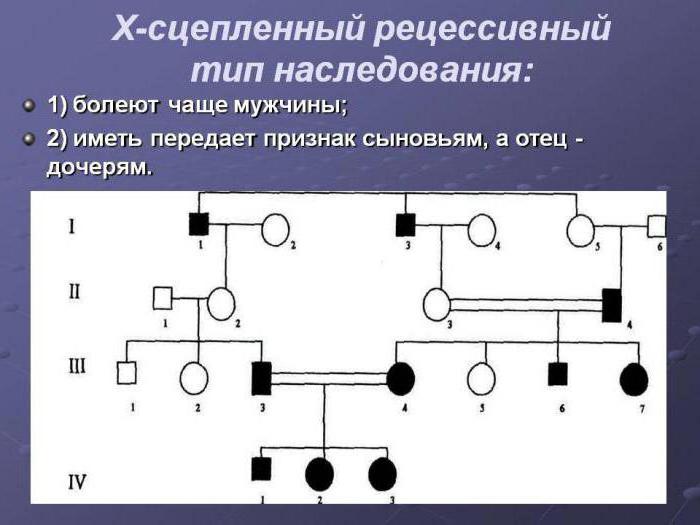
Х-ромосомный тип наследования у девочек и мальчиков проявляется по-разному. Это обусловлено наличием сразу двух Х-хромосом у женщины и одной у мужчины. Женский пол получает свои хромосомы по одной от каждого родителя, а мальчики – только от матери.
По этому типу наследования чаще всего патогенный материал передается женщинам, так как у них больше вероятности получить патогены от отца или матери. Если же носителем доминантного гена в семье является отец, то все мальчики будут здоровы, а у девочек будет проявляться патология.
При рецессивном типе Х-сцепления хромосом болезни проявляются у мальчиков с гемизиготным типом. Женщины всегда будут носителями больного гена, так как они гетерозиготные (в большинстве случаев), но если у женского пола будет гомозиготный признак, то она может заболеть.
Примером патологий с рецессивной Х-хромосомой могут быть: дальтонизм, дистрофия, болезнь Хантера, гемофилия.
Митохондриальный тип
Данный тип наследования является относительно новым. Митохондрии передаются с цитоплазмой яйцеклетки, в которых более 20 000 митохондрий. Каждая из них содержит хромосому. При данном типе наследования патологии передаются только по материнской линии. От таких матерей все дети рождаются больными.
При проявлении митохондриального признака наследственности у мужчин рождаются здоровые дети, так как этот ген не может передаваться от отца к ребенку, поскольку в спермии нет митохондрий.
Источник
Аутосомно-рецессивные заболевания проявляются только у гомозигот, которые получили по одному рецессивному гену от каждого из родителей. Заболевание может повторяться у сибсов пробанда, но иногда встречается и в боковых ветвях родословной. Характерным для аутосомно-рецессивных заболеваний является брак типа Аа х Аа (оба родителя здоровы, но являются носителями мутантного гена) (рис. 1Х.8).

Вероятность рождения больного ребенка (аа) в браке двух гетерозиготных носителей мутантного гена составляет 25%. Дети с рецессивными заболеваниями имеют, как правило, фенотипически здоровых родителей, и только после рождения больного ребенка ретроспективно можно установить генотип родителей и определить прогноз для будущих детей. Пример родословной с аутосомно-рецессивным типом наследования патологии приведен на рис. 1Х.9.
В популяции встреча двух носителей редкого аутосомно-рецессивного гена — нечастое событие, однако вероятность его значительно возрастает в случае родства супругов. Именно поэтому рецессивные заболевания часто проявляются в кровнородственных браках (рис. IX.10). По аутосомно-рецессивному типу наследуется абсолютное большинство наследственных заболеваний обмена веществ (ферментопатий). Наиболее частыми и значимыми в клиническом отношении являются такие болезни с аутосомно-рецессивным типом наследования, как муковисцидоз (кистофиб роз поджелудочной железы), фенилкетонурия, адреногенитальный синдром, многие формы нарушения слуха или зрения, болезни накопления.
На сегодняшний день известно более 1600 аутосомно-рецессивных заболеваний. Основные методы их предупреждения — медико-генетическое консультирование семей и дородовая диагностика (в случае заболеваний, для которых разработаны методы внутриутробной диагностики). Аутосомно-рецессивные болезни формируют значительную часть сегрегационно-


го генетического груза за счет высокой частоты патологического аллеля в популяции (табл. IX.2).

Для возникновения редких аутосомно-рецессивных заболеваний характерны следующие условия:
1) родители больного ребенка, как правило, здоровы;
2) мальчики и девочки заболевают одинаково часто;
3) повторный риск рождения ребенка с аутосомно-рецессивным заболеванием составляет 25%;
4) отмечается «горизонтальное» распределение больных в родословной, т. е. пациенты чаще встречаются в рамках одного сибства;
5) наблюдается увеличение частоты больных детей в группах родителей, связанных родством, причем чем реже аутосомно-рецессивное заболевание встречается в популяции, тем чаще больные происходят из кровнородственных браков.
- ПРОСТОЙ АУТОСОМНЫЙ РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ
- АУТОСОМНЫЙ ДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ
- АУТОСОМНО-РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ
- Аутосомно-рецессивные болезни
- СЦЕПЛЕННЫЙ С Х-ХРОМОСОМОЙ ТИП НАСЛЕДОВАНИЯ
- Х-СЦЕПЛЕННЫЕ РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ
- АУТОСОМНО-ДОМИНАНТНЫЕ ЗАБОЛЕВАНИЯ
- КРИТЕРИИ НАСЛЕДОВАНИЯ МОНОГЕННЫХ ЗАБОЛЕВАНИЙ
- Болезни, обусловленные иммунными комплексами (3-й тип иммунопатологических реакций). Характеристика иммунокомплексного воспаления. Нозологические формы заболеваний.
- Тип книдоспоридии и тип микроспоридии.
- АУТОСОМНО
- Аутосомно-доминантные моногенные болезни
- Х-СЦЕПЛЕННОЕ НАСЛЕДОВАНИЕ
- Другие аутосомные хромосомные болезни
- НАСЛЕДОВАНИЕ ПРИЗНАКОВ, СЦЕПЛЕННЫХ С ПОЛОМ
- МУЛЬТИФАКТОРИАЛЬНОЕ НАСЛЕДОВАНИЕ
- СЦЕПЛЕННОЕ НАСЛЕДОВАНИЕ ПРИЗНАКОВ
Источник
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Введение:
Аутосомно-рецессивное наследование — свойственный диплойдным эукариотам тип наследования признака, контролируемого рецессивным аллеям аутосомного гена. Для проявления мутации или болезни с таким типом наследования мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Иными словами, мутация проявляется только в гомозиготном состоянии, то есть тогда, когда обе копии гена, расположенные на гомологичных аутосомах, являются повреждёнными. Если мутация находится в гетерозиготном состоянии, и мутантному аллелю сопутствует нормальный функциональный аллель, то аутосомно-рецессивная мутация не проявляется.
Во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то по законам Менделя вероятность того, что дети, как и их родители, будут носителями мутантного гена составляет 50 %, вероятность рождения ребёнка без мутации — 25 %, вероятность рождения больного ребёнка — 25 %. При этом генные болезни этого типа наследования с одинаковой частотой встречаются как у мужчин, так и у женщин.
В основе многих наследственных заболеваний человека лежит данный тип наследования. Например, большинство лизосомных болезней накопления, к которым относятся болезнь Тея -Сакса, болезнь — Гоше, болезнь Ниманна — Пика и другие, являются аутосомно-рецессивными. Заболевание с аутосомно-рецессивным типом наследования клинически выражено только в случае, когда обе аутосомы являются дефектными по данному гену.
Распространённость болезней, наследуемых по аутосомно-рецессивному типу, зависит от частоты встречаемости рецессивного аллеля в популяции. Наиболее часто рецессивные наследственные болезни встречаются в изолированных этнических группах, а также среди населения с высоким процентом близкородственных браков.
Диггве-Мельхиор-Клаусена синдром.
Описан в 1962 г. H. Dyggve с соавт. Минимальные диагностические признаки:короткое туловище, скелетные аномалии.
Клиническая характеристика
Типичными признаками синдрома являются низкий рост, короткое туловище, выступающая грудина, бочкообразная грудная клетка, сколиоз, поясничный лордоз, ограничение подвижности суставов, маленькие кисти и стопы, утиная походка, микроцефалия, умственная отсталость. Рентгенологически определяются платиспондилия, широкие и короткие подвздошные кости, укорочение трубчатых костей с асимметричной оссификацией эпифизов и метафизов.
Типичны проксимальные концы бедренных костей— медиальная часть шейки бедра выступает в виде шпоры, ростковая зона расположена горизонтально, головка бедра оссифицируется поздно. Популяционная частота неизвестна.
Соотношения полов — M1:Ж1. Тип наследования— аутосомно-рецессивный. Дифференциальный диагноз:мукополисахаридоз, тип V, другие спондилоэпифизарные дисплазии.
Синдром Мардена-Уокера.
Синдром Мардена-Уокера – редкое заболевание соединительной ткани, которое наследуется по аутосомно-рецессивному признаку. Пациенты с этим расстройством, как правило, имеют отчетливое выражение лица, заячью губу или высокое арочное небо, маленькую челюсть, суставы в фиксированном положении, задержку роста и ограниченный контроль в движениях мышц. Синдром Мардена-Уокера развивается у мужчин чаще, чем у женщин.
Эпидемиология
Синдром Мардена-Уокера является очень редким заболеванием, которое развивается у мужчин чаще, чем у женщин с соотношением 11 к 3. В медицинской литературе описано около двадцати случаев.
Причины
Синдром Мардена-Уокера наследуется по аутосомно-рецессивному признаку. Точная генетическая аномалия пока не установлена.
Симптомы и проявления
Пациенты с синдромом Мардена-Уокера имеют различные черты лица, включая аномалии в челюсти, висящие веки, плоскую переносицу, низкие уши и выражение лица в фиксированном положении. Другие проявления этого расстройства включают искривление позвоночника в результате чего развивается горб, контрактуры суставов, расселины или высокое арочное небо, задержку роста и медленное движение мышц. Другие симптомы синдрома Мардена-Уокера могут включать в себя небольшую окружность головы, аномалии сердца, аномалии в мочевыделительной системе, снижение костной массы, аномально маленькие глаза, короткую шею, маленький рот и/или низкую линию роста волос. У небольшого количества пациентов с этим состоянием также могут иметь дополнительные ткани вызывающие обструкцию тонкой кишки, сужение кольца, отделяющего желудок от первой части тонкой кишки, что вызывает закупорку потока частично переваренной пищи (стеноз привратника); и/или потерю аппетита, неспособность организма усваивать питательные вещества должным образом, боли в желудке и потерю веса.
Девятимесячный мальчик с синдромом Мардена-Уокера. Тяжелые соматическое и психомоторное развитие, вес 2,5 кг, высота 55 см, окружность головы 43 см, гипотония, высунутый язык, полу-открытые глаза, контрактуры суставов, арахнодактилия и двусторонние паховые грыжи
Диагностика
Диагноз синдрома Мардена-Уокера ставится на основе минимальных критериев (задержка роста, тяжелая задержка развития, контрактуры суставов, блефарофимоз, микрогнатия, высокое арочное нёбо, низко посаженные уши и сколиоз). Магнитно-резонансная томография (МРТ) головы и эхокардиография могут помочь в выявлении пороков развития головного мозга и сердечно-сосудистых аномалий, соответственно. Кифоз диагностируется с помощью рентгена.
Лечение
Генетическое консультирование может быть полезным для пациентов и их семей. Лечение только симптоматическое и поддерживающее.
Синдром Маринеску-Шегрена
Синдром Маринеску-Шегрена – редкое наследственное заболевание с аутосомно-рецессивным типом наследования, характеризующееся триадой:
врождённая двусторонняя катаракта,
олигофрения,
спинно-мозжечковая атаксия.
Синдром впервые описан румынским невропатологом Г. Маринеску в 1931 году и шведским психиатром и невропатологом Шегреном в 1935 году. Всего описано около 60 случаев синдрома Маринеску-Шегрена. Для передачи по наследству имеет значение кровное родство родителей.
Одинаково часто заболевают мужчины и женщины.
Патоморфология
Патоморфологические изменения нервной системы при синдроме Маринеску-Шегрена неспецифичны:
в коре головного мозга отмечается атрофия ганглиозных клеток и разрежение миелинизированных волокон,
в мозжечке – массивная атрофия коры и дезинтеграция грушевидных нейроцитов (клеток Пуркинье).
Клиническая картина
Синдром Маринеску-Шегрена проявляется в раннем возрасте. Возникает слепота вследствие двусторонней катаракты, нарушается координация движений, позднее выявляется отставание в умственном развитии.
Непостоянными признаками являются:
низкий рост,
аномалии скелета (искривление позвоночника, микроцефалия, долихоцефалия),
слабость мышц конечностей,
пирамидные симптомы,
сходящееся косоглазие,
нистагм и др.
Течение заболевания медленно прогрессирующее.
Лечение
Лечение синдрома Маринеску-Шегрена симптоматическое.
Синдром Смита-Лемли-Опитца
Синдром Смита-Лемли-Опитца – комплекс аномалий развития многих органов и систем с нарушением физического и умственного развития. Р.
Частота:
1:20 000 новорожденных, соотношение полов М3:Ж1.
Минимальные диагностические признаки:
Микроцефалия, вывернутые наружу ноздри, птоз, синдактилия, гипоспадия и крипторхизм, умственная отсталость.
Клиника:
Низкие масса и длина тела при рождении (100%), микроцефалия, скафоцефалия, долихоцефалия, узкий лоб, деформированные и низко расположенные ушные раковины, птоз, эпикант, страбизм, короткий нос с широким кончиком и вывернутыми наружу ноздрями (100%), длинный фильтр, микрогнатия и широкий альвеолярный край верхней челюсти (100%), расщелина неба. Конечности – кожная (редко костная) синдактилия стоп, постаксиальная полидактилия кистей и/или стоп, косолапость, вывих бедра, клинодактилия, флексорное положение пальцев рук, поперечная ладонная складка. У мальчиков – гипоспадия и крипторхизм, у девочек – гипертрофия клитора. Сосковый гипертелоризм, дистальный акроцианоз. Внутренние органы – врожденные пороки сердца (сужение устья аорты, стеноз легочной артерии), аномалии почек (поликистоз, гидронефроз, удвоение лоханок, аномалии мочеточников), аномалии лобуляции легких, пилоростеноз, паховые грыжи, гипоплазия тимуса. Умственная отсталость (100%), рвота (90%), судороги.
Патологическая анатомия:
Головной мозг – расширение желудочков, гипоплазия и агенезия мозолистого тела, гипоплазия лобных долей, липома гипофиза, гипоплазия мозжечка с гипоплазией червя, голопорэнцефалия. Нарушение миграции нейронов, обширный глиоз, эктопия клеток Пуркинье.
Лечение:
Симптоматическое.
Прогноз:
Относительно благоприятный при отсутствии пороков внутренних органов.
Синдром Шегрена-Ларссона.
Определение синдрома Шегрена-Ларссона. Эндемически встречающаяся в Швеции наследственная форма олигофрении в комбинации с церебральной диплегией и врожденным ихтиозом.
Авторы.
Sjogren Karl Gustaf Torsten — современный шведский психиатр, Стокгольм. Larsson Tage — современный шведский психиатр, Стокгольм. Впервые синдром описали в 1957 г. Sjogren и Larsson.
Симптоматология синдрома Шегрена-Ларссона:
1. Олигофрения всех степеней.
2. Спастическая диплегия, тип Little (S. Little) с симметричным распространением, особенно выраженная на верхних конечностях; иногда отмечают прогрессирование спазмов.
3. Врожденный универсальный ихтиоз.
4. В отдельных случаях одновременно имеется также дегенерация сетчатой оболочки глаза в области maculae с резко выраженным снижением зрения.
5. Некоторые авторы к проявлениям этого синдрома относят также карликовый или гигантский рост, гипоплазию половых органов, пернициозоподобную анемию [S. (Pseudo) Biermer], причем чаще в таких случаях говорят о «S. Rud», хотя в случаях, наблюдавшихся Rud, не было ни идиотии, ни спастической диплегии.
Этиология и патогенез.
Моногибридное аутосомно-рецессивное наследственное страдание. Заболевание встречается и впервые было описано в северных районах шведской провинции Вестерботтен. При так называемом S. Rud, по-видимому, имеет место гипофизарное расстройство.
Дифференциальный диагноз. S. Little (см.). Детский мозговой паралич. Изолированный врожденный ихтиоз. S. de Sanctis—Cacchione (см.).
ЛИТЕРАТУРА.
1. Афонькин С.Ю. Секреты наследственности человека. –С-Пб.: «Корона-Принт». 2002, –352 с.
2. Бадалян Л.О. «Наследственные болезни у детей» — М., 1971.
3. Леруа Арман Мари. Мутанты. О генетической изменчивости и человеческом теле. -М.: Изд-во «АCT», 2010. –560 с
4. Бочков Н.П. «Генетика человека (Наследственность и
патология)» – М., 1978
5. Козлова С.И. «Наследственные синдромы и медико-генетическое
консультирование» – М., 1996.
6. Боринская С.А., Янковский Н.К. Люди и их гены -Фрязино: “Век 2”. 2005. –64 с.
Источник