Болезни связанные с нарушениями в системе репарации

РЕПАРАЦИЯ — функция клеток, заключающаяся в способности исправлять повреждения в молекулах ДНК (была обнаружена в 1949 г. А. Кельнером, Р. Дюльбекко и И.Ф. Ковалевым независимо друг от друга).
ФОТОРЕАКТИВАЦИЯ или СВЕТОВАЯ репарация. В результате УФ – облучения целостность молекул ДНК нарушается, так как в ней возникают димеры, т. е. сцепленные между собой соединения в области пиримидиновых оснований. Фотореактивация катализируется ферментом фотолиазой, который активируется фотоном света и расщепляет димер на исходные составляющие.
ТЕМНОВАЯ или ЭКСЦИЗИОННАЯ репарация. Осуществляется в пять этапов: 1 – нарушения узнаются специфическими белками; 2 – эндонуклеазы делают надрезы в поврежденной цепи; 3 – экзонуклеазы осуществляют вырезание поврежденного участка; 4 – синтез нового участка по принципу комплементарности взамен удаленного фрагмента, с помощью ДНК-полимеразы; 5 – ДНК-лигаза соединяет концы старой цепи и восстановленного участка.
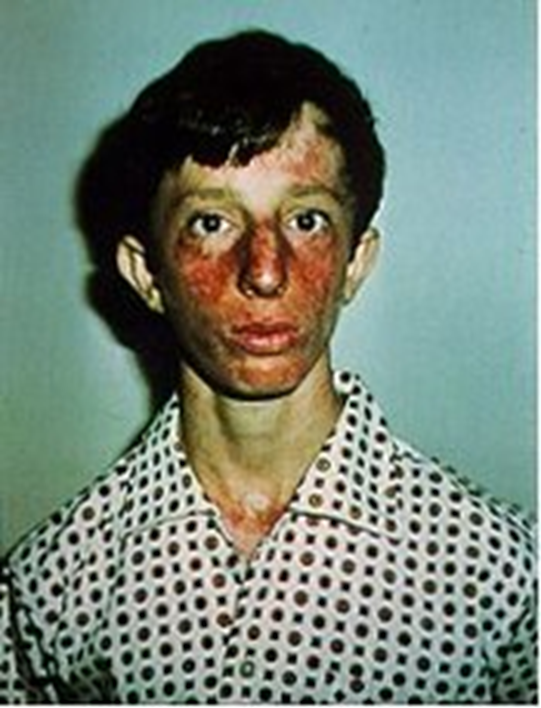
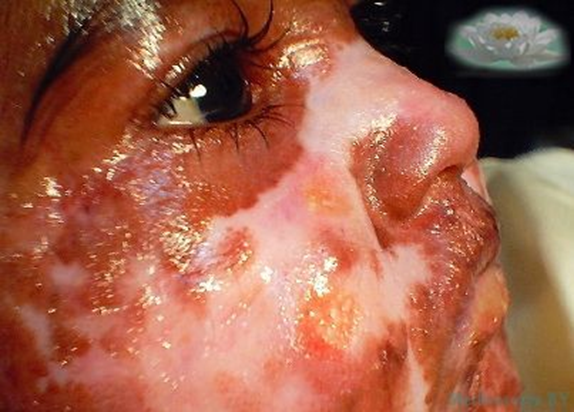 С нарушением репаративных процессов в ДНК связано развитие некоторых заболеваний. Пигментная ксеродерма –наследственное заболевание кожи, проявляющееся повышенной чувствительностью к ультрафиолетовому облучению, проявляется в возрасте двух-трех лет. В результате мутации неактивны белки, репарирующие ДНК. Повреждения накапливаются и со временем приводят к раку кожи.
С нарушением репаративных процессов в ДНК связано развитие некоторых заболеваний. Пигментная ксеродерма –наследственное заболевание кожи, проявляющееся повышенной чувствительностью к ультрафиолетовому облучению, проявляется в возрасте двух-трех лет. В результате мутации неактивны белки, репарирующие ДНК. Повреждения накапливаются и со временем приводят к раку кожи.
Синдром Блума –аутосомно-рецессивное заболевание, характеризующееся телеангиэктатической эритемой лица, повышенной фоточувствительностью, задержкой роста. Культура лимфоцитов и фибробластов больных отличается высокой частотой хромосомных аберраций и повышенной чувствительностью к УФ-облучению, отмечена недостаточность фермента ДНК-лигазы I.
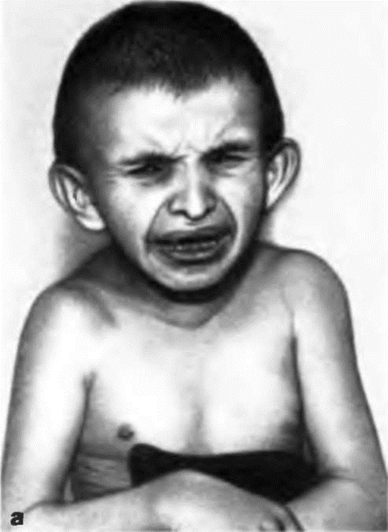 Синдром Коккейна – наследственное заболевание, с поражением кожи и её придатков, органов зрения, слуха и нарушением репарации. Синдром обусловлен мутациями в генах, кодирующие белки, ответственные за эксцизионную репарацию. Ребенок имеет внешний вид «старика», волосы быстро седеют, повышенная фоточувствительность может стать причиной появления на лице пигментации.
Синдром Коккейна – наследственное заболевание, с поражением кожи и её придатков, органов зрения, слуха и нарушением репарации. Синдром обусловлен мутациями в генах, кодирующие белки, ответственные за эксцизионную репарацию. Ребенок имеет внешний вид «старика», волосы быстро седеют, повышенная фоточувствительность может стать причиной появления на лице пигментации.
Анемия Фанкони – аутосомно-рецессивное заболевание, характеризующееся поражением всех элементов костного мозга, наблюдается нарушение вырезания пиримидиновых димеров, а также нарушение репарации межцепочечных сшивок.
2.9. Генетическая инженерия — совокупность приёмов, методов и технологий получения рекомбинантных РНК и ДНК, выделения генов из организма (клеток), осуществления манипуляций с генами и введения их в другие организмы. Основными направлениями генной инженерии являются: создание трансгенных растений и животных, разработка принципов генной терапии.
Генно-инженерные работы выполняются в несколько этапов:
1. Получают нужный ген, готовый для последующих процедур трансгенеза. Ген может быть выделен из естественных источников, синтезирован химически по имеющейся последовательности нуклеотидов, получен с помощью полимеразной цепной реакции или путем копирования ДНК на РНК-матрице с использованием обратной транскриптазы;
2. Подбирают вектор, обладающий всеми необходимыми характеристиками;
3. Вектор и клонированный ген обрабатывают одинаковыми рестриктазами;
4. Сшивают вектор и встроенный ген с помощью ДНК-лигазы;
5. Вводят рекомбинантную конструкцию из вектора и встроенного гена в клетки–мишени реципиента – осуществляют трансформации;
6. Проверяют наличие трансгена в клетках – мишенях.
Источник
Для любых клеток проблема защиты своей ДНК от всевозможных повреждений является настолько фундаментальной, что ответственные за подобную протекцию гены оказались очень схожими и у человека, и у грызунов, и у пивных дрожжей. Не вызывает сомнения, что эти гены являются неотъемлемой частью генома практически любого организма. Нарушения в работе этих важных генов приводят к тяжелейшим последствиям. У человека наиболее известные генетические заболевания – пигментная ксеродерма, синдром Коккейна и трихотиодистрофия.
В 1968 г. англ. ученым Д. Кливером было доказано, что наследственная болезнь человека — пигментная ксеродермия. Ксеродерма распространена во всем мире. По счастью, частота ее встречаемости невелика и составляет, например, в США 1 случай на 250 тыс., а в Японии – на 40 тыс. Больные ксеродермой (от греч. «ксеро» – сухой и «дерма» – кожа) крайне чувствительны к солнечному свету. На поверхности их кожи часто возникают множественные веснушки и даже рубцы, кожа меняет свою пигментацию и становится сухой. При этом заболевании страдают прежде всего ткани и органы, на которые непосредственно воздействует солнечный свет, в частности, роговица, сетчатка и хрусталик глаза. Самое печальное то, что у больных часто уже в юном возрасте возникают различные виды рака кожи – меланомы и карциномы. Средний возраст больных ксеродермой, у которых появляются раковые опухоли, составляет всего 8 лет, тогда как в среднем подобные новообразования появляются только к 50 годам. Даже в полости рта, куда свет попадает все же нечасто, у больных ксеродермой опухоли возникают в 20 тыс. раз чаще.
Ксеродерма – наследственное рецессивное не связанное с полом заболеванием. Это означает, что она с равной вероятностью встречается как у женщин, так и у мужчин и проявляется только в том случае, если нарушения в соответствующих генах присутствуют сразу в двух хромосомах – одной материнской и парной ей отцовской.
Что именно повреждается в системе репарации ДНК при ксеродерме, еще не до конца ясно. Известно лишь, что клетки таких больных сверхчувствительны к воздействию УФ-лучей, химических мутагенов и канцерогенных веществ, образующих с ДНК прочные соединения. Вместе с тем эти клетки обычно сохраняют нормальную чувствительность к ионизирующей радиации.
Как это нередко бывает в случае генетически обусловленных заболеваний, ксеродерма является следствием повреждения не одного участка ДНК, а нескольких. К настоящему времени выделяют по меньшей мере семь типов повреждений и, как следствие, семь различных форм этого заболевания.
В дальнейшем было установлено, что еще некоторые наследственные болезни человека обусловлены нарушениями процессов генетической репарации. К числу этих заболеваний относится синдром Хатчинсона, при котором развивается карликовость, преждевременное старение и прогрессирующее слабоумие. Повреждением генов, кодирующих ферменты репарации, обусловлено возникновение ряда форм такой относительно распространенной болезни, как системная красная волчанка.
Трихотиодистрофией называется еще одно генетическое врожденное рецессивное заболевание человека, связанное с нарушением репарации ДНК, хотя эта связь менее очевидна, чем в случае пигментной ксеродермы.
Если поместить волосы больных трихотиодистрофией под микроскоп, становятся видны характерные поперечные полосы, возникающие из-за нехватки в них богатых серой белков. Пучок таких волос становится ломким и напоминает по окраске тигриный хвост. Под таким названием («тигриный хвост») трихотиодистрофия и вошла в историю медицины в 1968 г. К сожалению, это не единственные признаки заболевания. Значительно серьезнее то, что оно связано с нарушением интеллектуальных функций, маленьким ростом и пониженной плодовитостью. У некоторых больных трихотиодистрофией наблюдается также повышенная чувствительность к УФ-облучению, что прямо указывает на повреждение систем репарации ДНК. Вместе с тем у таких больных нет предрасположенности к раку кожи.
Синдром Коккейна. Повышенной фоточувствительностью страдают также люди с редким заболеванием, которое было описано в средине 1930-х гг. Коккейном и получило название по фамилии этого исследователя. На генетическую рецессивную природу синдрома указывали его проявления в семьях с родственными браками. Помимо гиперчувствительности к свету больные синдромом Коккейна страдают также глухотой и атрофией зрительного нерва. У них укороченное туловище, большие уши и нос, а также глубоко запавшие глаза. В довершение ко всему они страдают от кариеса и стареют несколько быстрее, чем остальные люди. У них наблюдается ряд мозговых нарушений. Редко кто из людей с таким диагнозом доживает до 18–20 лет.
Клетки соединительной ткани людей с синдромом Коккейна очень плохо выживают после таких доз УФ-облучения, которые для клеток здоровых людей являются относительно безвредными. Этот факт указывает на то, что все описанные выше неприятные проявления болезни могут быть косвенными следствиями нарушения репарации ДНК.
Специалисты могут назвать еще несколько врожденных заболеваний человека, которые, скорее всего, вызваны нарушениями репарации ДНК. Среди них атаксия-телеангиэктазия, при которой рождаются люди с так называемой атаксией – прогрессивным церебральным параличом и патологическим расширением кровеносных сосудов на коже и конъюнктиве глаз. Они страдают умственной и иммунной недостаточностью. Приблизительно у 10 % больных в раннем возрасте развиваются злокачественные опухоли. По оценкам специалистов каждый сотый, а по некоторым данным даже каждый двадцатый человек является носителем гена этого заболевания, которое, по счастью, не проявляется, если в гомологичной хромосоме работает соответствующий неповрежденный ген.
У больных синдромом Блума, еще одним наследственным заболеванием, связанным с нарушением репарации ДНК, обнаружена недостаточность ДНК-лигазы. Вследствие этого у них повышена вероятность возникновения онкогенных заболеваний.
Ясно, что подобные генетические нарушения систем контроля за состоянием ДНК должны выбраковываться естественным отбором и поэтому не закрепляются в человеческой популяции. Те немногие случаи, которые все-таки удается обнаружить, позволяют ученым изучать тонкие механизмы репарации ДНК в клетках человека и те страшные последствия, к которым приводят нарушения в их работе.
Изучение молекулярной природы этих заболеваний дает основание надеяться на относительно быструю разработку методов их лечения. Успехи в этом направлении зависят как от исследования деталей процессов генетической репарации и изучения возможности выделения из нормальных организмов (в особенности микробов) активно работающих ферментов с последующим введением их в организм больного, так и от методов замещения больных генов здоровыми. Если второй путь пока остается только в области гипотез, то в первом направлении начата экспериментальная работа. Так, японские исследователи К. Танака, М. Бекгучи и И. Окада в конце 1975 г. сообщили об успешном использовании одного из репарирующих ферментов, выделенных из клеток бактерий, зараженных бактериальным вирусом, для устранения дефекта в клетках, взятых от больного, страдающего пигментной ксеродермией. Для того чтобы этот фермент мог успешно проникнуть в клетки человека, культивировавшиеся в искусственных условиях, был использован убитый вирус Сендай. Однако до настоящего времени подобные работы не удается проводить на организме человека. Другое направление связано с разработкой способов ранней диагностики болезней, обусловленных дефектами репарирующих ферментов.
Проектные задания
1. Составьте сводную таблицу «Типы мутаций»:
2. Дайте характеристику следующим синдромам в электронном виде: — синдром Дауна; — синдром Лежена; — синдром Шерешевского-Тернера; — синдром Патау; — синдром Эдвардса. Подберите фотографии, иллюстрирующие клинические проявления данных синдромов, и изображения кариотипов больных. 3. Выберите 10 типов мутаций и запишите их по правилам современной номенклатуры мутаций. 4. Опишите любой из известных вам метод обнаружения геномных мутаций. В электронном виде последовательно отобразите этапы проведения, выбранного вами метода обнаружения мутаций. 5. Напишите реферат «Репарация ДНК у человека». Рекомендуемые страницы: Воспользуйтесь поиском по сайту: ©2015- 2020 megalektsii.ru Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. |
Источник
![]()
Íàñëåäñòâåííûå çàáîëåâàíèÿ, õàðàêòåðèñòèêà èõ êëèíè÷åñêèõ ïðîÿâëåíèé. Îñîáåííîñòè ýòèîëîãèè è ïàòîãåíåçà áîëåçíåé, ñâÿçàííûõ ñ ãåíåòè÷åñêèìè íàðóøåíèÿìè. Àóòîñîìíî-ðåöåññèâíûé òèï íàñëåäîâàíèÿ. Ïðè÷èíà ñèíäðîìà ïðåæäåâðåìåííîãî ñòàðåíèÿ (ïðîãåðèè).
| Ðóáðèêà | Ìåäèöèíà |
| Âèä | ðåôåðàò |
| ßçûê | ðóññêèé |
| Äàòà äîáàâëåíèÿ | 21.12.2015 |
| Ðàçìåð ôàéëà | 48,5 K |
Îòïðàâèòü ñâîþ õîðîøóþ ðàáîòó â áàçó çíàíèé ïðîñòî. Èñïîëüçóéòå ôîðìó, ðàñïîëîæåííóþ íèæå
Ñòóäåíòû, àñïèðàíòû, ìîëîäûå ó÷åíûå, èñïîëüçóþùèå áàçó çíàíèé â ñâîåé ó÷åáå è ðàáîòå, áóäóò âàì î÷åíü áëàãîäàðíû.
Ðàçìåùåíî íà https://www.allbest.ru/
ïðîãåðèÿ ñèíäðîì íàñëåäñòâåííîå çàáîëåâàíèå
Ãåíåòèêà
Àíåìèÿ Ôàíêîíè â áîëüøèíñòâå ñëó÷àåâ èìååò àóòîñîìíî-ðåöåññèâíûé òèï íàñëåäîâàíèÿ. Ýòî îçíà÷àåò, ÷òî äâå êîïèè ãåíà (îäíà îò ìàòåðè è îäíà îò îòöà) íåñóò â ñåáå ìóòàöèè. Ïðè ýòîì âåðîÿòíîñòü ðîæäåíèÿ áîëüíîãî ðåáåíêà ó òàêèõ ðîäèòåëåé ñîñòàâëÿåò 25 %.  ðåäêèõ ñëó÷àÿõ àíåìèÿ Ôàíêîíè èìååò Õ-ñöåïëåííûé ðåöåññèâíûé òèï íàñëåäîâàíèÿ. Ïðè ýòîì òîëüêî ìàòü ÿâëÿåòñÿ íîñèòåëåì ìóòàöèè, à âåðîÿòíîñòü çàáîëåâàíèÿ ó ñûíîâåé ñîñòàâëÿåò 50 %.
 íàñòîÿùåå âðåìÿ èçâåñòíî 15 ãåíîâ, ñâÿçàííûõ ñ àíåìèåé Ôàíêîíè. Òîëüêî îäèí èç íèõ — FANCB — íàõîäèòñÿ íà Õ-õðîìîñîìå. Îñòàëüíûå ãåíû ðàñïîëîæåíû íà àóòîñîìàõ. Êàæäûé èç ýòèõ ãåíîâ îòâå÷àåò çà ñèíòåç îïðåäåë¸ííîãî ôåðìåíòà. Êëåòêè îò äâóõ áîëüíûõ, âûðàùåííûå â êëåòî÷íîé êóëüòóðå â ëàáîðàòîðíûõ óñëîâèÿõ, ìîãóò äîïîëíÿòü äðóã äðóãà è ôóíêöèîíèðîâàòü íîðìàëüíî, åñëè îíè íåñóò íàðóøåíèÿ â ðàçíûõ ãåíàõ. Ïðè íàëè÷èè ìóòàöèé â îäíîì è òîì æå ãåíå ýòîãî íå ïðîèñõîäèò, è êëåòêè íå äîïîëíÿþò äðóã äðóãà. Ïî ýòîìó ïðèíöèïó âûäåëÿþò ãðóïïû êîìïëèìåíòàöèè: A, B, C, D , êàæäîé èç êîòîðûõ ñîîòâåòñòâóåò îïðåäåëåííûé ãåí: FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCP è RAD51C.  êàæäîé ýòíè÷åñêîé ãðóïïå åñòü ìóòàöèè, õàðàêòåðíûå äëÿ íå¸. Òàê ñðåäè àøêåíàçîâ ÷àùå âñåãî âñòðå÷àåòñÿ ìóòàöèÿ â ãåíå FANCC.
Ðàçìåùåíî íà Allbest.ru
…
Ïîäîáíûå äîêóìåíòû
Íàñëåäñòâåííûå çàáîëåâàíèÿ ÷åëîâåêà. Àóòîñîìíî-ðåöåññèâíûé òèï íàñëåäîâàíèÿ. Ïîíÿòèå âðîæäåííîé äåôîðìàöèè. Ãëèîìà ñåò÷àòêè ãëàçà. Àóòîñîìíî-äîìèíàíòíîå íàñëåäîâàíèå àíîìàëèé. Ïèãìåíòíàÿ äèñòðîôèÿ ñåò÷àòêè. Íàñëåäñòâåííûå àòðîôèè çðèòåëüíîãî íåðâà.
ïðåçåíòàöèÿ [1,8 M], äîáàâëåí 07.12.2016
Êëàññèôèêàöèÿ íàñëåäñòâåííûõ áîëåçíåé ÷åëîâåêà. Ãåííûå, ìèòîõîíäðèàëüíûå è õðîìîñîìíûå áîëåçíè. Ïîâðåæäåíèÿ íàñëåäñòâåííîãî àïïàðàòà êëåòêè. Îáùàÿ ÷àñòîòà ãåííûõ áîëåçíåé â ïîïóëÿöèÿõ ëþäåé. Ïðèçíàêè ñèíäðîìà Ìàðôàíà è ìåòîäû ëå÷åíèÿ ãåìîôèëèè.
ïðåçåíòàöèÿ [2,5 M], äîáàâëåí 06.12.2012
Ïîíÿòèå è ñóùíîñòü áèîëîãè÷åñêîãî è ïàñïîðòíîãî âîçðàñòà ÷åëîâåêà. Ôèçèîëîãèÿ è àíàëèç ñòàðåíèÿ îðãàíèçìà. Îñîáåííîñòè ïðîÿâëåíèÿ è òå÷åíèÿ áîëåçíåé ó ñòàðèêîâ ïî Í.Ä. Ñòðàæåñêî. Ñðàâíèòåëüíàÿ õàðàêòåðèñòèêà ïðåæäåâðåìåííîãî è ôèçèîëîãè÷åñêîãî ñòàðåíèÿ.
êîíòðîëüíàÿ ðàáîòà [25,7 K], äîáàâëåí 07.04.2010
Ãåííûå áîëåçíè, ñâÿçàííûå ñ ìóòàöèÿìè îòäåëüíûõ ãåíîâ çà ñ÷åò ïðåîáðàçîâàíèé õèìè÷åñêîé ñòðóêòóðû ÄÍÊ. Ïðè÷èíû âîçíèêíîâåíèÿ, ïàòîãåíåç áîëåçíåé îáìåíà âåùåñòâ. Ïðîòåêàíèå è ýòèîëîãèÿ çàáîëåâàíèé, ñöåïëåííûõ ñ ïîëîì. Âðîæäåííûå çàáîëåâàíèÿ ó äåòåé.
ïðåçåíòàöèÿ [9,6 M], äîáàâëåí 14.03.2013
Êëàññèôèêàöèÿ è äèôôåðåíöèàöèÿ íàñëåäñòâåííûõ çàáîëåâàíèé. Ãåííûå è õðîìîñîìíûå áîëåçíè, áîëåçíè ñ íàñëåäñòâåííîé ïðåäðàñïîëîæåííîñòüþ. Ãåíåòè÷åñêèå êàðòû ÷åëîâåêà, ëå÷åíèå è ïðåäóïðåæäåíèå íåêîòîðûõ íàñëåäñòâåííûõ áîëåçíåé. Îïèñàíèå îñíîâíûõ áîëåçíåé.
ïðåçåíòàöèÿ [1,4 M], äîáàâëåí 16.11.2011
Íàñëåäñòâåííûå áîëåçíè, îáóñëîâëåííûå õðîìîñîìíûìè è ãåííûìè ìóòàöèÿìè. Ôàêòîðû ðèñêà íàñëåäñòâåííîãî çàáîëåâàíèÿ. Ïðîôèëàêòèêà è ìåäèêî-ãåíåòè÷åñêîå êîíñóëüòèðîâàíèå. Ñèìïòîìàòè÷åñêîå ëå÷åíèå íàñëåäñòâåííûõ áîëåçíåé. Êîððåêöèÿ ãåíåòè÷åñêîãî äåôåêòà.
ïðåçåíòàöèÿ [1,4 M], äîáàâëåí 03.12.2015
Ñóùíîñòü ïîíÿòèÿ «íàñëåäñòâåííûå çàáîëåâàíèÿ». Ìíîãîãåííûå, õðîìîñîìíûå, ïîëèãåííûå íàñëåäñòâåííûå áîëåçíè. Ãðóïïû õðîìîñîìíûõ áîëåçíåé: àíîìàëèè ÷èñëà õðîìîñîì, íàðóøåíèÿ ñòðóêòóðû. Ñèíäðîì Äàóíà, Ïàòó. Ãåíåòè÷åñêèå áîëåçíè ñîìàòè÷åñêèõ êëåòîê.
ïðåçåíòàöèÿ [556,1 K], äîáàâëåí 06.04.2011
Èññëåäîâàíèå ìåõàíèçìà ðàçâèòèÿ, ýòèîëîãèè è ïàòîãåíåçà áûñòðîïðîãðåññèðóþùåãî ãëîìåðóëîíåôðèòà. Õàðàêòåðèñòèêà êëèíè÷åñêèõ ïðîÿâëåíèé, ìîðôîëîãè÷åñêèõ èçìåíåíèé â êëóáî÷êàõ ïî÷åê. Îñîáåííîñòè äèàãíîñòèêè, ìåòîäû ëå÷åíèÿ è ïðîãíîç çàáîëåâàíèÿ ó äåòåé.
ïðåçåíòàöèÿ [565,8 K], äîáàâëåí 07.12.2012
Îïðåäåëåíèå ïîíÿòèÿ áðîíõîîáñòðóêòèâíîãî ñèíäðîìà. Îïèñàíèå åãî ýòèîëîãèè, ïàòîãåíåçà, êëèíè÷åñêèõ ñèìïòîìîâ, èñòî÷íèêîâ, ôàêòîðîâ ðèñêà, îñíîâíûõ ìåòîäîâ äèàãíîñòèêè è ëå÷åíèÿ çàáîëåâàíèÿ. Îñîáåííîñòè ëå÷åíèÿ áðîíõîîáñòðóêòèâíîãî ñèíäðîìà ó äåòåé.
ïðåçåíòàöèÿ [3,4 M], äîáàâëåí 30.09.2017
Ñòðîåíèå óõà ÷åëîâåêà. Êàê ïðîèñõîäèò çâóêîâîñïðèÿòèå. Îïèñàíèå ýòèîëîãèè, ïàòîãåíåçà, êëèíè÷åñêèõ ñèìïòîìîâ è ìåòîäîâ ëå÷åíèÿ çàáîëåâàíèé óõà: îòîñêëåðîçà, áîëåçíè Ìåíüåðà, ïîâðåæäåíèÿ áàðàáàííîé ïåðåïîíêè, íàðóæíîãî è ñðåäíåãî îòèòà, ëàáèðèíòà.
ðåôåðàò [1,5 M], äîáàâëåí 28.03.2019
- ãëàâíàÿ
- ðóáðèêè
- ïî àëôàâèòó
- âåðíóòüñÿ â íà÷àëî ñòðàíèöû
- âåðíóòüñÿ ê íà÷àëó òåêñòà
- âåðíóòüñÿ ê ïîäîáíûì ðàáîòàì
Источник
Ксеродемрма пигмемнтная (синонимы: ретикулярный прогрессирующий меланоз, прогрессирующий ретикулярный меланоз Пика) — наследственное заболевание кожи, проявляющееся повышенной чувствительностью к ультрафиолетовому облучению, проявляется в возрасте двух-трех лет и постоянно прогрессирует. Является предраковым состоянием кожи. Встречается редко.
Имеет значение наследственный фактор. Характер наследственного дефекта заключается в отсутствии или малой активности ферментов, устраняющих повреждающий эффект ультрафиолетового излучения на клетки кожи. В результате мутации белки, репарирующие ДНК больного, становятся неактивны, и при всяком повреждении, например, при облучении ультрафиолетом, дефектных молекул ДНК становится больше. Повреждения накапливаются и со временем приводят к раку кожи. Изучены два вида нарушений. При одном из них, помимо высокой чувствительности к УФ-излучению, у больных имеет место и повышенная чувствительность к радиации. Результатом в обоих случаях являются нарушения пигментации и ороговения кожи, атрофические изменения эпидермиса и дистрофия соединительнотканных волокон, а конечным эффектом — клеточная атипия и озлокачествление.
Атаксия телеангиэктазия (англ. Ataxia Telangiectasia) (АТ), известная также как синдром Луи-Бара (англ. Louis-Bar syndrome) является редким нейродегенеративным наследственным заболеванием, вызывающим тяжёлую инвалидность. Атаксия приводит к плохой координации, а телеангиэктазия — слегка расширенным кровеносным сосудам; оба признака являются отличительными чертами болезни. Атаксия телеангиэктазия вызывается мутациями вгене ATM, который отвечает за ответ клетки на двунитевые разрывы в ДНК.
При атаксии телеангиэктазии страдают различные органы и функции организма. Во-первых, при этом заболевании ухудшается работа определённых областей мозга, включая мозжечок, что вызывает трудности с движениями и координацией. Во-вторых, при АТ ослаблена иммунная система, что приводит к предрасположенности к инфекции. В-третьих, при АТ нарушена репарация ДНК, что увеличивает риск развития рака.
Симптомы чаще всего появляются в раннем детстве, когда дети начинают ходить. Хотя больные АТ, как правило, начинают ходить в нормальном возрасте, они раскачиваются или колеблются при ходьбе, стоя или сидя, и может показаться, что они пьяны. В конце дошкольного и младшего школьного возраста у них развиваются трудности с перемещением глаз из одного направления в другое (глазодвигательная апраксия). У них развивается невнятная или искажённая речь и глотания. Некоторые из них имеют повышенную инфекционную восприимчивость дыхательных путей (инфекции уха, синусит, бронхит и пневмония). Поскольку не все дети развиваются с одинаковой скоростью, диагностика заболевания может занять несколько лет. Большинство детей с AT имеют стабильные неврологические симптомы в течение первых 4-5 лет жизни, но ещё бульшее число проблем у них возникает в младшем школьном возрасте.
Синдром Блума или Синдром Блум-Торре-Мачэйкик — редкое аутосомно-рецессивное заболевание, для которого характерны низкий рост больных и предрасположенность к онкологическим заболеваниям. Клетки больных синдромом Блума демонстрируют сильную геномную нестабильность, в частности, частую гомологичную рекомбинацию.
Синдром Блума — разновидность прогероидных синдромов и демонстрирует ряд признаков, характерных для этой группы заболеваний: короткий рост и рано развивающаяся сыпь на областях кожи, контактирующих с солнечным светом. Кожная сыпь эритемная, склеродермическая и масштабная. При поражении кожи в области носа и щек, сыпь имеет бабочкообразную форму.
Белок синдрома Блума — белок, кодируемый у людей геном BLM, и который не экспрессируется в случае наличия синдрома Блума.
Продукт гена BLM связан с подмножеством DExH box-содержащих хеликаз и имеет как ДНК-стимулированную АТФазную активность, так и АТФ-зависимую ДНК хеликазную активность. Мутации, вызывающие синдром Блума, изменяют или приводят к удалению хеликазного мотива и могут полностью убрать 3′ > 5′ хеликазную активность. В норме этот белок может выступать супрессором неверной гомологичной рекомбинации.
Трихотиодистрофия (TTD) — аутосомно-рецессивное наследственное заболевание, характеризующееся ломким волосом и интеллектуальными нарушениями. TTD связана с диапазоном симптомов, связанных с органами эктодермы и нейроэктодермы. TTD могут быть подразделены на четыре синдрома: Примерно половина всех пациентов с трихотиодистрофией страдают светочувствительностью, которая делит классификацию синдромов с ней или без нее; BIDS и PBIDS, IBIDS и PIBIDS. Современное использование включает TTD-P (светочувствительная) и TTD.
Особенности TTD могут включать светочувствительность, ихтиоз, ломкие волосы и ногти, интеллектуальные нарушения, снижение рождаемости и низкий рост. Аббревиатуры PIBIDS, IBIDS, BIDS и PBIDS представляют инициалы участвующих слов. Синдром BIDS , является аутосомно-рецессивным наследственным заболеванием. Это касается не светочувствительных синдромов. Существует светочувствительный синдром, PBIDS.
BIDS связан с геном MPLKIP (TTDN1) .
Все светочувствительные синдромы TTD имеют дефекты в их системах NER. NER служит для нуклеотидной эксцизионной репарации, которая является жизненной системой репарации ДНК, устраняющей многие виды повреждений ДНК. Этот дефект не присутствует в не светочувствительных TTD. Дефекты NER могут привести к другим редким аутосомно-рецессивным заболеваниям, как пигментная ксеродерма и синдром Коккэйна.
Синдром Коккейна (англ. Cockayne syndrome, CS), также называемый синдром Нил-Дингуолл (англ. Neill-Dingwall)) — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемыхлейкодистрофия (состояние, характеризующееся деградацией белого вещества). В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо поняты.
Лица с этим синдромом имеют меньший обычного размер головы (микроцефалия), невысокий рост (карликовость), глаза выглядят запавшими, и они имеют «пожилой» вид. Они часто имеют длинные конечности с контрактурами суставов (неспособностью расслабления мышцы в суставе), сутулость (кифоз) и они могут быть очень тощими (кахексия) из-за потери подкожного жира. Их маленький подбородок, большие уши, и заостренный, тонкий нос часто дают пожилой внешний вид. Кожа лиц с синдромом Коккейна также часто аномальна. Гиперпигментация, варикозные или узловатые вены (телеангиэктазия) и серьезная чувствительность к солнечному свету являются общими симптомами, даже у лиц без XP-CS. Часто пациенты с синдромом Коккейн бывают слабо чувствительны к ожогам и волдырям. Глаза пациентов могут быть затронуты различными путями и их аномалии распространены в CS. Катаракта и помутнение роговицы являются общими симптомами.
Прогеримя (др. — греч. рспу — сверх, гЭсщн — старик) — один из редчайших генетических дефектов. При прогерии возникают изменения кожи и внутренних органов, которые обусловлены преждевременным старением организма. Классифицируют детскую прогерию (синдром Гетчинсона (Хатчинсона) — Гилфорда) и прогерию взрослых (синдром Вернера).
В мире зафиксировано не более 80 случаев прогерии Причина детской прогерии — мутации гена LMNA, кодирующего ламин А. Ламины — белки, из которых выстроен особый слой оболочки клеточного ядра. В большинстве случаев прогерия встречается спорадически, в нескольких семьях зарегистрирована у сибсов, в том числе от кровнородственных браков, что свидетельствует о возможности аутосомно-рецессивного типа наследования. В клетках кожи больных обнаружены нарушения репарации ДНК и клонирования фибробластов, а также атрофические изменения эпидермиса и дермы, исчезновение подкожной клетчатки.
Прогерия взрослых имеет аутосомно-рецессивный тип наследования. Дефектный ген — WRN (ген АТФ-зависимой хеликазы). Предполагается связь процесса с нарушением репарации ДНК, обмена соединительной ткани.
Синдром Вернера впервые был описан немецким врачом Отто Вернером в 1904 году, но до сих пор прогерия остается малоизученным заболеванием, прежде всего, из-за редкой встречаемости. Известно, что это генетическое нарушение, вызванное мутацией генов, которое передается по наследству.
На сегодняшнее время ученые определили также, что синдром Вернера является аутосомно-рецессивным заболеванием. Это значит, что больные прогерией получают одновременно от отца и матери по одному аномальному гену, находящемуся в восьмой хромосоме. Однако до сих пор подтвердить или опровергнуть диагноз посредством генетического анализа не представляется возможным.
Источник