Хромосомные и генные мутации наследственных болезни

По изменению генетического материала выделяют три группы мутаций: генные, хромосомные и геномные.
Генными, или точечными (точковыми), называют мутации, возникающие в результате изменения гена, т. е. структуры молекулы ДНК.
При нарушении репликации может произойти изменение последовательности нуклеотидов в каком-нибудь участке ДНК. Это может быть:
- замена нуклеотида;
- вставка нуклеотида;
- выпадение нуклеотида.
Если происходит замена нуклеотида, то результат может быть разный. В некоторых случаях такая мутация не приводит к изменению структуры белка.
Пример:
рассмотрим мутацию ГТТ ЦЦЦ ГГТ → ГТЦ ЦЦЦ ГГТ.
В первом триплете произошла замена тимина на цитозин. Триплеты ГТТ и ГТЦ кодируют глутаминовую кислоту, поэтому никаких изменений в структуре белка данная мутация не вызывает: глу-гли-про → глу-гли-про.
В других случаях замена нуклеотида может изменить порядок аминокислот в молекуле белка и привести к фенотипическим последствиям.
Пример:
мутация ГТТ ЦЦЦ ГГТ → ГТГ ЦЦЦ ГГТ.
В первом триплете произошла замена тимина на гуанин. Триплет ГТТ кодирует глутаминовую кислоту, а триплет ГТГ — гистидин. Значит, первичная структура белка изменяется: глу-гли-про → гис-гли-про. Это может привести к фенотипическим изменениям.
Добавление или выпадение нуклеотидов приводит к сдвигу рамки считывания в рибосоме и к изменению последовательности аминокислот. Синтезируется белок, который отличается своей первичной структурой от исходного. В результате может произойти серьёзное изменение фенотипа.
Пример:
ГТТ ЦЦЦ ГГТ Т → ГТЦ ЦЦГ ГТТ.
Исходный участок ДНК кодирует аминокислотную последовательность глу-гли-про. После выпадения тимина в первом нуклеотиде последовательность аминокислот другая: лиз-глу-глу. Мутагенный ген передаёт к месту синтеза новую информацию, синтезируется другой белок, что может привести к возникновению нового признака.
Генные мутации приводят к таким наследственным заболеваниям, как фенилкетонурия (нарушение обмена веществ) и альбинизм (отсутствие нормальной пигментации).
Хромосомными называют мутации, обусловленные изменением структуры хромосом.
Это может быть:
- утрата (нехватка) — потеря хромосомой своей концевой части;
- делеция — выпадение участка средней части хромосомы;
- дупликация — удвоение фрагмента хромосомы;
- инверсия — поворот участка хромосомы на (180)°;
- транслокация — перенос участка одной хромосомы на другую.
Хромосомные мутации чаще всего возникают при нарушении деления клеток. Их последствия для организма могут быть разными. Наиболее опасны утрата и делеция, так как может быть потеряна информация о жизненно важном белке.
Нарушение структуры хромосом у человека часто приводит к тяжёлым формам умственной отсталости, заболеваниям крови, снижению жизнеспособности организма.
Пример:
потеря небольшой части (21)-й хромосомы вызывает лейкоз.
Хромосомные мутации можно обнаружить с помощью микроскопа. Микроскопирование используется в диагностике наследственных заболеваний.
Геномными называют мутации, обусловленные изменением числа хромосом в кариотипе организма.
Различают полиплоидию и анеуплоидию (гетероплоидию).
Полиплоидия — кратное увеличение гаплоидного набора хромосом.
Возникает при нарушении расхождения хромосом при митозе или мейозе.
В результате хромосомный набор клетки становится триплоидным 3n, тетраплоидным 4n, гексаплоидным 6n и т. д.
Полиплоидия широко используется в селекции растений. Полиплоидные растения, как правило, характеризуются более мощным ростом, большей продуктивностью, жизнеспособностью. Для получения полиплоидных растений используют колхицин, который разрушает нити веретена деления и приводит к образованию полиплоидных геномов.
Диплоидное растение
Полиплоидное растение
Анеуплоидия (гетероплоидия) — некратное изменение числа хромосом 2n±1, 2n±2…
Этот вид мутаций может быть обусловлен избытком или недостатком одной или нескольких хромосом. Причиной гетероплоидии является нарушение расхождения гомологичных хромосом при мейозе. В одну гамету попадают обе гомологичные хромосомы, а в другую — ни одной. Слияние такой гаметы с нормальной и приводит к образованию зиготы с большим или меньшим числом хромосом по сравнению с исходным хромосомным набором.
Различают следующие формы анеуплоидии:
- трисомия (2n+1) — три хромосомы в одной из пар (трисомия по (21)-й паре хромосом у человека — синдром Дауна);
- моносомия (2n−1) — недостаток одной хромосомы (моносомия по X-хромосоме — синдром Шерешевского-Тернера);
- нуллисомия (2n−2) — отсутствие пары гомологичных хромосом (летальная мутация).
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 12 марта 2020;
проверки требует 1 правка.
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны (известно свыше 6000) и разнообразны по проявлениям.
Некоторые наследственные заболевания являются врождёнными. Врождённые наследственные болезни следует отличать от пороков развития, вызванных, например, инфекцией (сифилис или токсоплазмоз) или воздействием иных повреждающих факторов на плод во время беременности.
Причина заболеваний и классификация[править | править код]
При наследственных заболеваниях могут иметь место генетические нарушения различного характера и локализации. Эти болезни могут быть связаны с нарушениями ядерной (хромосомной) или митохондриальной ДНК. Они могут развиться в результате генных (точечных) мутаций (транзиции, трансверсии[1], мутации сдвига рамки считывания[2][* 1]), либо довольно грубых изменений структуры хромосом[3] или мтДНК[4] (делеции, дупликации, инверсии, транслокации, транспозиции[5]), а также вследствие геномных мутаций (изменения числа хромосом[6]). Соответственно, наследственные заболевания классифицируют как генные, хромосомные, митохондриальные.
Наследственные заболевания классифицируют также по типу наследования. Для значительной части наследственных болезней тип наследования установлен — патологические признаки, также как и нормальные, могут наследоваться аутосомно-доминантно, аутосомно-рецессивно и сцепленно с полом (Х-сцепленный доминантный, Х-сцепленный рецессивный и Y-сцепленный типы наследования). Термин «аутосомный» указывает на то, что мутантный ген локализован в аутосоме, «Х-сцепленный» — в половой Х-хромосоме, а «Y-сцепленный» — в половой Y-хромосоме. Выделение доминантного и рецессивного типов наследования существенно с медицинской точки зрения, так как при доминантном типе наследования клиническое проявление болезни обнаруживается у гомо- и гетерозигот, а при рецессивном — только у гомозигот, то есть значительно реже. Основные методы, с помощью которых устанавливается тот или иной тип наследования, — клинико-генеалогический, базирующийся на анализе родословных, и более точный сегрегационный анализ, объектом которого, как правило, являются так называемые «ядерные семьи» (то есть родители и дети).
Болезни, обусловленные дефектами ядерной ДНК
- Генные болезни, связанные с точечными мутациями ядерной ДНК:
- Моногенные заболевания, связанные с мутациями ядерной ДНК;
- Полигенные болезни, связанные с мутациями ядерной ДНК.
- Болезни экспансии тринуклеотидных повторов.
- Хромосомные болезни
- Связанные с изменением структуры хромосом[* 2].
- Связанные с изменением числа хромосом[* 3]:
Болезни, обусловленные дефектами митохондриальной ДНК
- Генные болезни, связанные с точечными мутациями мтДНК.
- Болезни, обусловленные грубыми структурными нарушениями мтДНК.
Наследственные заболевания, обусловленные дефектами ядерной ДНК[править | править код]
Традиционно выделяют три подгруппы заболеваний, связанных с мутациями ядерной ДНК: моногенные наследственные заболевания, полигенные наследственные болезни и хромосомные болезни. Первые две группы обусловлены точечными мутациями ДНК (генные болезни), третья группа соответствует грубым структурным перестройкам хромосом (хромосомным аберрациям[5]) или изменению их числа.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое, порой весьма долгое, время. Так, при хорее Гентингтона дефектный ген обычно проявляет себя только на третьем-четвёртом десятилетии жизни, проявление признаков спинальной мускульной атрофии (СМА) наблюдается в возрасте от 6 месяцев до 4—50 лет (в зависимости от формы заболевания).
Генные болезни, связанные с точечными мутациями ядерной ДНК[править | править код]
Это наиболее широкая группа наследственных заболеваний. В настоящее время описано более 4000 вариантов моногенных наследственных болезней, подавляющее большинство которых встречается довольно редко (например, частота серповидноклеточной анемии — 1/6000).
Широкий круг моногенных болезней образуют наследственные нарушения обмена веществ, возникновение которых связано с мутацией генов, контролирующий синтез ферментов и обусловливающих их дефицит или дефект строения — ферментопатии.
У точечных мутаций ядерной ДНК может быть один из трёх типов наследования: аутосомно-доминантный, аутосомно-рецессивный и сцепленное с полом наследование. Тип наследования мутации можно определить при помощи генеалогического исследования.
Полигенные наследственные болезни[править | править код]
Полигенные болезни наследуются сложно. Для них вопрос о наследовании не может быть решён на основании законов Менделя. Ранее такие наследственные заболевания характеризовались как болезни с наследственной предрасположенностью. Однако сейчас о них идёт речь как о мультифакториальных заболеваниях с аддитивно-полигенным наследованием с пороговым эффектом.
К этим заболеваниям относятся такие болезни как рак, сахарный диабет, шизофрения, эпилепсия, ишемическая болезнь сердца, гипертензия и многие другие.
Хромосомные болезни[править | править код]
Хромосомные болезни обусловлены грубыми нарушениями наследственного аппарата — изменением числа[* 4] или структуры[* 5]хромосом[7].
Сюда относятся синдромы Дауна, Клайнфельтера, Шерешевского — Тернера, Эдвардса, «кошачьего крика» и другие. (См. также Радиационная генетика)
Болезни, обусловленные дефектами митохондриальной ДНК[править | править код]
Митохондриальная ДНК (мтДНК) наследуется по материнской линии[8]. Патологические нарушения клеточного энергетического обмена, обусловленные мутациями мтДНК, могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Вследствие гетероплазмии проявление и степень тяжести болезней, обусловленных сходными нарушениями мтДНК, могут быть неодинаковыми у разных людей[9], в зависимости от соотношения в цитоплазме клеток мутантных и нормальных митохондрий[4].
Генные болезни, связанные с точечными мутациями мтДНК[править | править код]
Имеется ряд моногенных болезней, связанных с точечными дефектами митохондриальных генов:
- наследственная оптическая нейропатия Лебера[* 6] обусловлена миссенс-мутацией в одном из восемнадцати генов, кодирующих белки окислительного фосфорилирования[4];
- болезнь миоклональной эпилепсии и грубо-красных волокон[4][* 7] (MERRF) — вызвана точечной заменой в гене тРНК[4][9];
- синдром митохондриальной энцефаломиопатии и инсультоподобных эпизодов (MELAS) — также развивается вследствие точечной замены в гене тРНК[9].
Болезни, обусловленные грубыми структурными нарушениями[править | править код]
К числу болезней, связанных с грубыми перестройками мтДНК, относятся:
- синдром Кернса—Сэйра[* 8], обусловленный делециями больших участков мтДНК[4];
- синдром Пирсона[11].
Диагностика[править | править код]
Диагностика производится цитогенетическими, молекулярно-цитогенетическими и молекулярно-генетическими методами, а также на основании фенотипических признаков (синдромологический подход) и биохимических показателей.
Статистика[править | править код]
Статистика заболеваний, вызванных генетическими нарушениями.
Примерно 5-6 детей из 100 рождаются с какими-нибудь генетически обусловленными заболеваниями. В большинстве — это заболевания с генетическими предрасположенностями. Это могут быть пороки развития, нарушения в интеллектуальном развитии ребёнка. В эти 5-6 процентов входят наследственные заболевания, возникшие впервые или унаследованные от одного из родителей.
— И. Наумчик[12].
Примечания[править | править код]
Комментарии
- ↑ Вследствие вставки или выпадения нуклеотидов[1].
- ↑ Именно такие перестройки ядерной ДНК С. Г. Инге-Вечтомов называет хромосомными мутациями, или хромосомными аберрациями[6].
- ↑ Геномными мутациями[6].
- ↑ Геномные мутации.
- ↑ Хромосомные мутации.
- ↑ Другое название болезни — атрофия дисков зрительных нервов Лебера[10].
- ↑ Другое название болезни — синдром миоклонус-эпилепсии и рваных красных мышечных волокон[9].
- ↑ Другое название — синдром Кернса—Сайра[9].
Источники
- ↑ 1 2 Инге-Вечтомов, 1989, с. 308.
- ↑ Марри и др., 1993, с. 98—100.
- ↑ Жимулёв, 2002, с. 51.
- ↑ 1 2 3 4 5 6 Жимулёв, 2002, с. 454.
- ↑ 1 2 Инге-Вечтомов, 1989, с. 318—346.
- ↑ 1 2 3 Инге-Вечтомов, 1989, с. 293.
- ↑ Инге-Вечтомов, 1989, с. 516.
- ↑ Жимулёв, 2002, с. 455.
- ↑ 1 2 3 4 5 Гинтер, 2003, с. 130.
- ↑ Гинтер, 2003, с. 129.
- ↑ Гинтер, 2003, с. 130—131.
- ↑ Замдиректора РНПЦ «Мать и дитя»: Примерно 5-6 детей из 100 рождаются с генетически обусловленными заболеваниями. news.tut.by (24 марта 2009). Дата обращения 16 мая 2013. Архивировано 20 мая 2013 года.
Литература[править | править код]
- Гинтер Е. К. Медицинская генетика. — М.: Медицина, 2003. — 448 с. — 5000 экз. — ISBN 5-225-04327-5.
- Жимулёв И. Ф. Общая и молекулярная генетика. — Новосибирск: Сиб. унив. изд-во, 2002. — 459 с. — 2000 экз. — ISBN 5-7615-0509-6.
- Инге-Вечтомов С. Г. Генетика с основами селекции. — М.: Высш. шк., 1989. — 591 с. — 21 000 экз. — ISBN 5-06-001146-1.
- Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. — М.: Мир, 1993. — Т. 2. — 415 с. — ISBN 5-03-001775-5.
См. также[править | править код]
Источник
В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний — бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) — врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента — меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.

Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.

Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя — третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией — трисомией по 18 паре хромосом. Причина — нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией — трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
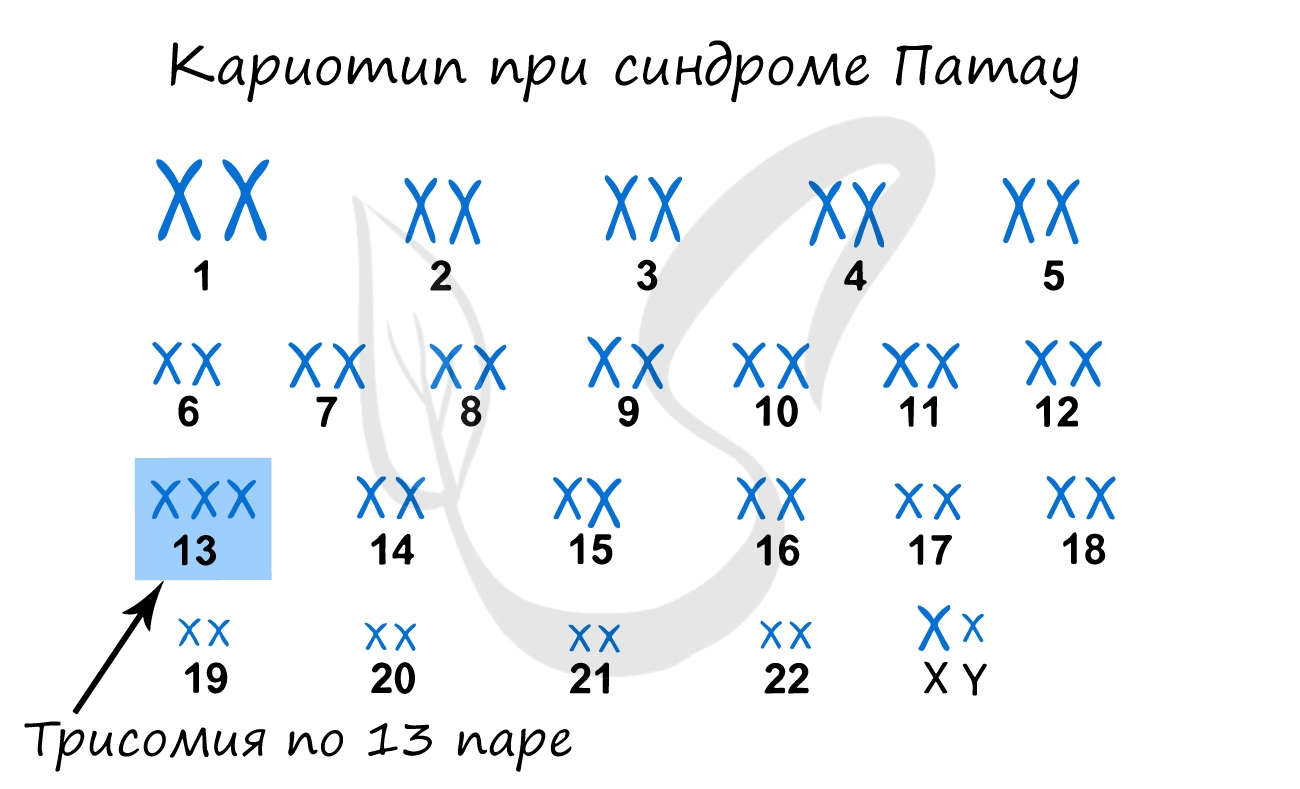
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

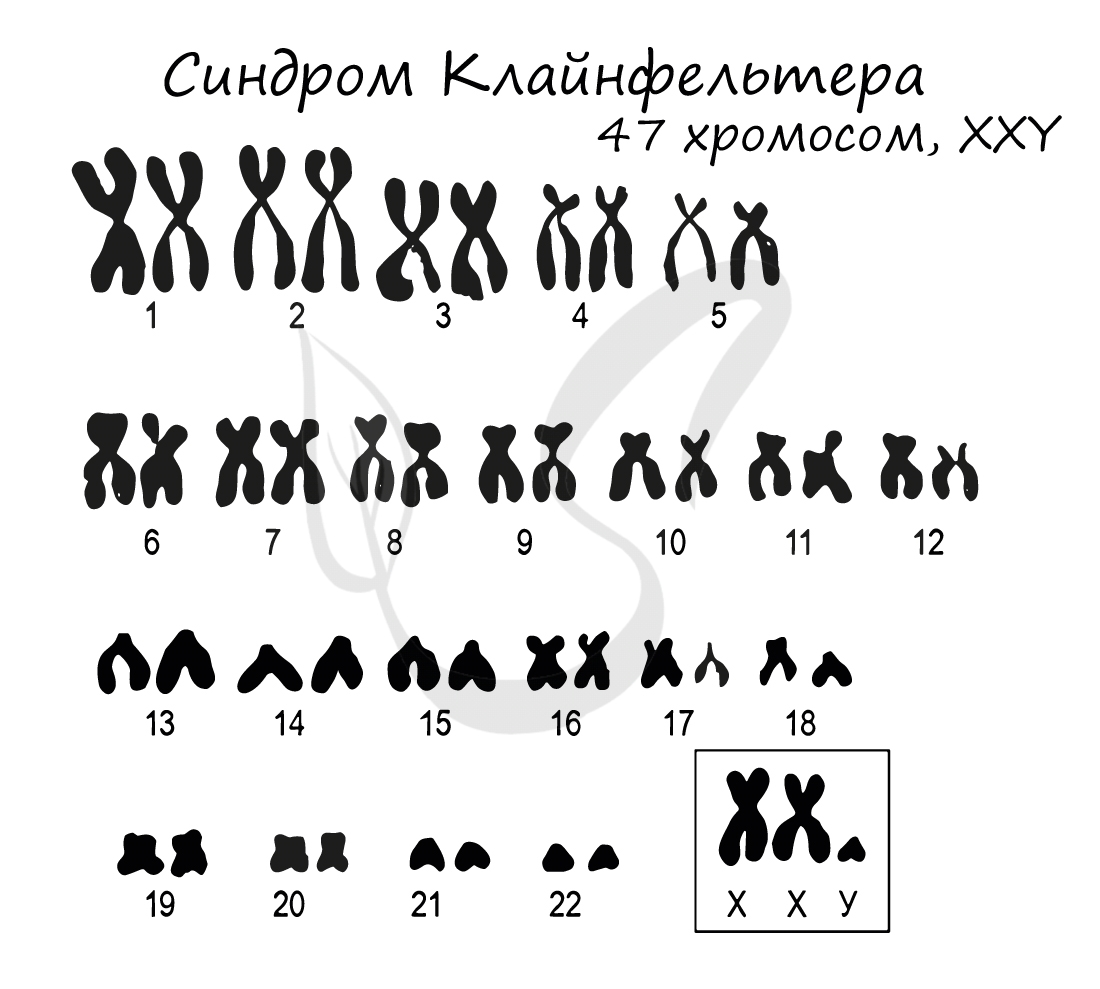
Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа — «XY» 🙂
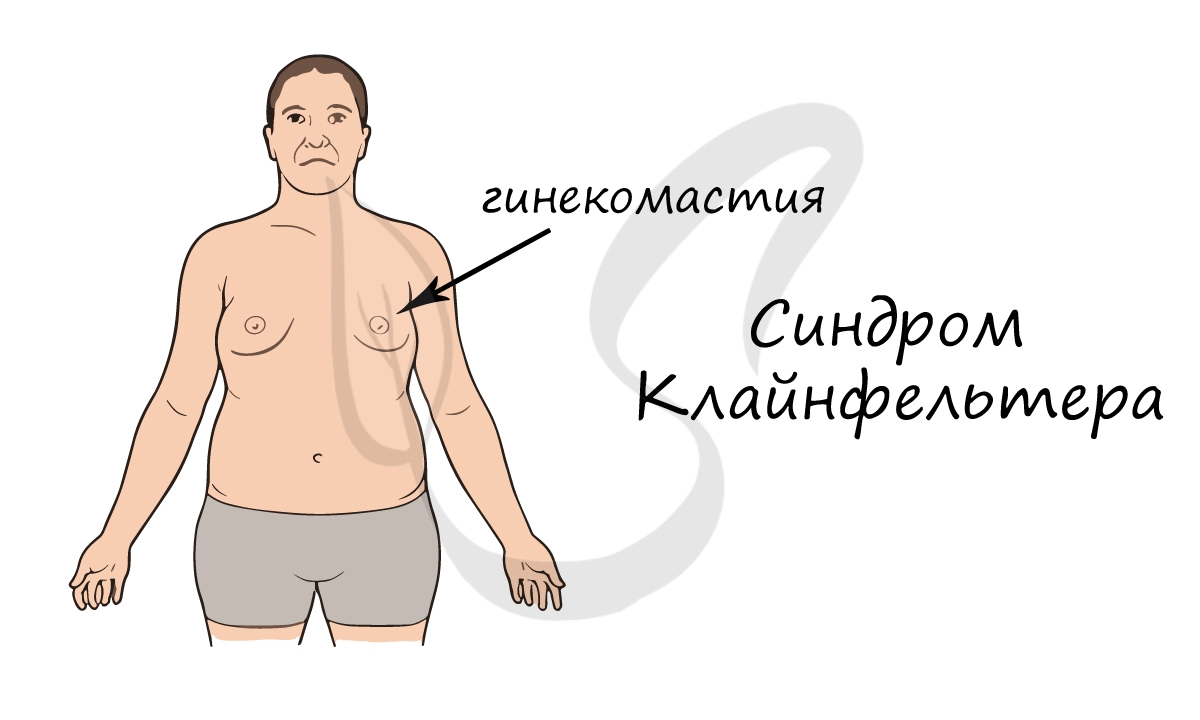
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера — наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения — эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2020
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник