Мутации в генах при болезни дауна

Почему у здоровых мужчин и женщин могут родиться дети с хромосомными мутациями? Какие болезни родственников должны стать причиной для обращения к генетику? Как составляется карта генетических рисков? На эти и другие вопросы Yellmed ответила врач-генетик сети центров репродукции и генетики Нова Клиник Анастасия Владимировна Волкова.
Фото: pixabay.com
– Анастасия Владимировна, как гены могут помешать женщине стать матерью, а мужчине – отцом?
– Чтобы ответить на этот вопрос, немного расскажу об организации генетического материала. Хромосомы расположены в ядре клетки, они сложены из непрерывной ДНК. И вот эту ДНК мы условно делим на гены.
На данный момент наиболее изучены генетические причины мужского бесплодия на уровне генов. У мужчин есть на У-хромосоме фактор азооспермии AZF, который состоит из 3 регионов: a, b, c. Выпадение или отсутствие части или полностью региона сопровождается нарушением созревания и деления сперматозоидов.
В некоторых случаях, чтобы «получить» сперматозоид для оплодотворения в программе ЭКО, необходимо провести биопсию яичка.
У мужчин также может быть нарушен отток спермы по семявыносящим канальцам. Это происходит либо из-за повышенной вязкости спермы, либо в силу недоразвития семявыносящих протоков. Такое патологическое состояние мы видим у мужчин, больных наследственным заболеванием – муковисцидозом, а также у носителей мутации в гене CFTR, ответственном за развитие данного заболевания.
– Часто будущие родители не подозревают, что являются носителями серьезных наследственных заболеваний или даже имеют хромосомные мутации. В какой момент проблема «вылезает» наружу?
– Если рассматривать целые хромосомы, состоящие из большого количества генов, то и здесь можно обнаружить причины, приводящие к бесплодию или рождению больного ребенка.
Речь идет о носителях сбалансированных перестроек в своих хромосомах. Внешне это здоровые люди, без клинических проявлений. Однако когда встает вопрос о деторождении, в семье наступает неразвивающаяся беременность, возможно, потом еще одна. Ко мне приходят семьи с двумя и тремя потерями беременности в анамнезе. При исследовании хромосом обоих супругов – анализ кариотипа – обнаруживается сбалансированная перестройка. Например, транслокация – обмен генетическим материалом между двумя хромосомами. При сбалансированном типе перестройки не происходит потери генов, поэтому внешне перестройка никак себя не проявляет. Но у носителя повышен риск передачи своему ребенку хромосом с утратой участка гена или дупликацией, то есть в несбалансированном виде.
Некоторые перестройки чаще приводят к выкидышам на ранних сроках, другие – к рождению ребенка с хромосомной аномалией. Трисомия 21-й хромосомы, или синдром Дауна, в ряде случаев «передается» от родителей-носителей. Бессимптомное носительство мутаций есть и на уровне генов.
Установлено, что каждый человек является носителем мутаций в своем геноме, иначе говоря, они находятся в спящем состоянии. Чтобы заболевание «проявилось», муж и жена должны иметь мутации в одном и том же гене.
Тогда одна дефектная копия гена передастся ребенку от папы, а вторая — от мамы. В этой ситуации ребенок будет болен. Данный тип наследования называется аутосомно-рецессивный, риск для потомства в семье двух носителей составит 25%.
– Как человеку догадаться, что он является носителем опасных болезней, связанных с патологиями генов?
– Генетик при планировании беременности рекомендует пройти генетическое тестирование на скрининг носительства мутаций в генах. Семья может выбрать расширенное обследование, например, поиск частых мутаций в генах самых распространенных аутосомно-рецессивных заболеваний. Выделяют также этноспецифический скрининг – когда мы знаем, что в данной популяции высокая частота носительства по конкретным заболеваниям. Например, у армян распространено наследственное заболевание – средиземноморская лихорадка, или периодическая болезнь. Каждый десятый является носителем мутации в гене, ответственном за развитие заболевания.
Фото: Нова Клиник
– Какие болезни родственников – родителей, бабушек и дедушек – должны стать сигналом, что нужно пройти консультацию генетика, прежде чем самому планировать детей?
– Всего описано около 7 тысяч редких болезней, и с каждым днем их количество увеличивается.
В первую очередь, семья должна обратиться на консультацию к генетику, если у ближайших родственников были случаи умственной отсталости, врожденных и наследственных заболеваний, расстройств аутистического спектра, эпилепсии, бесплодия, ранней младенческой смертности, у женщин – ранней менопаузы до 40 лет, у мужчин – шатающейся походки – атаксии, тремора после 45-50 лет.
– У моей мамы была СКВ – системная красная волчанка – серьезное и сложное аутоиммунное заболевание. Стоит ли мне беспокоиться и при планировании беременности сдавать специальные генетические анализы?
– Системная красная волчанка – многофакторное заболевание, то есть болезнь с наследственной предрасположенностью. Разовьется ли заболевание при жизни у человека, зависит от действия множества генов и наличия факторов внешней среды. Поэтому не имеет смысла сдавать генетические анализы.
– А если не знаешь, какое было здоровье у дальних родственников? Или, например, о твоем собственном отце ничего не известно, а о его родителях – и подавно.
– При отсутствии информации о здоровье ближайших родственников семья может пройти скрининг носительства мутаций, чтобы определить, какие мутации в «спящем» состоянии «достались» от родителей.
– Как составляется карта генетических рисков?
– Карта генетических рисков – это расширенная панель скрининга носительства мутаций в генах. Панели могут включать разное количество заболеваний. При составлении панели лаборатория руководствуется следующими критериями: высокая частота носительства в популяции, заболевания имеют четкие клинические проявления и оказывают пагубное влияние на качество жизни, требуют хирургического или медикаментозного вмешательства и прочие.
– Какие риски чаще всего обнаруживаются?
– Частота «обнаружения» зависит от частоты носительства мутаций. Наиболее распространены моногенные заболевания: спинальная амиотрофия, частота носительства в популяции, в среднем, 1:32 — 1:40, и муковисцидоз – 1:45.
– Если у супругов происходит конфликт на уровне хромосом и годами не наступает беременность, им можно как-то помочь? У них есть шанс стать родителями?
– Да, можно. В некоторых случаях у мужчин-носителей транслокаций выявляют выраженные изменения в показателях спермы. Естественным путем в такой ситуации беременность не наступит. Поэтому лечение бесплодия рекомендовано методом ЭКО с проведением ПГТ – преимплантационного генетического тестирования.
ПГТ необходимо для выявления у эмбриона несбалансированных перестроек и численных нарушений хромосом, которые может принести сперматозоид для оплодотворения.
Заранее определив «здоровые» эмбрионы, мы увеличиваем шанс наступления беременности.
– Как парам, планирующим беременность, узнать, что у них есть серьезные хромосомные мутации?
– Узнать о носительстве изменений, то есть перестроек, в своих хромосомах можно только путем исследования хромосом – кариотипирования. Для этого нужно всего лишь сдать анализ крови. Если же семья уже столкнулась с бесплодием, невынашиванием беременности, необходима консультация генетика. Задача врача – провести внешний осмотр супругов, собрать информацию об обследовании и лечении, составить родословную.
– В каких случаях назначают кариотипирование – микроскопическое исследование для выявления хромосомных аномалий?
– Кариотипирование, как тест первой линии, рекомендовано при: задержке полового развития, первичной или вторичной аменорее, ранней менопаузе, бесплодии неясной этиологии, невынашивании беременности, множественных пороках развития у плода, хромосомных аномалиях, выявленных при исследовании материла замершей беременности, хромосомной аномалии у ребенка, выраженном нарушении сперматогенеза, а также детям при задержке психо-речевого, моторного и физического развития, врожденных пороках развития, малых аномалиях развития, с неясным полом при рождении.
Хочу отметить, исследование хромосом в микроскоп – очень сложная и кропотливая работа.
– Как проводится анализ эмбриона на наличие генных отклонений? Говоря простым языком – как к нему подобраться?
– После оплодотворения образуется зигота, которая начинает активно делиться. На 5-6 сутки развития формируется бластоциста. Она представлена двумя рядами клеток и полостью.
Первый ряд клеток – это внутренняя клеточная масса, которая в дальнейшем даст развитие самому ребенку. Второй ряд клеток – трофэктодерма, из которой будет формироваться плацента. В большинстве случаев генетический материал одинаковый во внутренней клеточной массе и трофэктодерме. Эмбриолог проводит биопсию, то есть забор нескольких клеток от трофэктодермы. Эти клетки отправляют в лабораторию на исследование, сами эмбрионы замораживают.
Фото: pixabay.com
– Можно полностью застраховаться от всех «генетических сюрпризов»? Или есть патологии, которые проявляются только на этапе эмбрионального развития и предсказать их невозможно?
– Не существует всеобъемлющего исследования, которым можно исключить все мутации по разным группам заболеваний. Например, молекулярно-генетический метод исследования нового поколения NGS позволяет определить численные нарушения по всем 46 хромосомам, но микроскопические потери или удвоения участков хромосом посмотреть не удастся.
Проводя поиск генных мутаций, мы не обнаружим изменения числа хромосом – анеуплоидии. «Осечки» могут быть также и потому, что исследуется только несколько клеток трофэктодермы эмбриона, а для полноценного исследования требуется большее количество клеток.
К патологиям, которые проявляются только на этапе эмбрионального развития, можно отнести пороки развития плода. Какие-то из них будут иметь мультифакториальную природу, какие-то «возникнут» в результате тератогенного действия, например, инфекции – ветряной оспы, краснухи и других или радиационного облучения на ранних этапах развития ребенка.
– Почему у женщин зрелого возраста существенно повышается риск появления у эмбриона синдрома Дауна? И это правда, что после сорока лет он составляет 80%?
– Да, действительно, у женщин с 35 лет увеличивается риск рождения ребенка не только с синдромом Дауна, но другими численными хромосомными аномалиями, например, синдром Эдвардса и синдром Патау. Это связано с увеличением риска неправильного деления яйцеклетки. В результате яйцеклетка принесет для оплодотворения лишнюю хромосому по какой-то паре. При синдроме Дауна лишняя 21-я хромосома, при синдроме Эдвардса – 18-я хромосома, а при синдроме Патау – 13-я.
Если рассматривать программу ЭКО, то доля эмбрионов с правильным набором хромосом у женщин 40 лет и старше составляет примерно 20%. В отношении синдрома Дауна: в 40 лет риск родить ребенка с трисомией 21-й хромосомы составляет 1:63, а в 45 лет – 1:19.
– Женщины, которые хотят забеременеть после 40, пусть даже с помощью ЭКО, наверняка, понимают, на какой риск идут. Бывает так, что их этот риск останавливает?
– Не все понимают, в силу отсутствия знаний и информации о наследственной патологии. Осознание в большинстве случаев происходит на консультации генетика.
В программе ЭКО, даже получив полноценную информацию о риске генетической патологии, есть семьи, отказывающиеся от проведения преимплантационного генетического тестирования.
Причины разные: боязнь проведения биопсии клеток эмбриона для исследования, ведь у некоторых пар можно «получить» лишь один или 2 эмбриона, религиозные убеждения и другие обстоятельства.
– Какой процент риска рождения ребенка с наследственной или врожденной патологией говорит о том, что целесообразно прервать беременность?
– Процент риска ни в одной ситуации не является показанием для прерывания беременности. Для выявления генетической патологии необходимо проведение подтверждающего метода – инвазивной пренатальной диагностики с целью забора материала и генетического исследования.
При подтверждении генетической патологии, например трисомии 21 хромосомы, семья может прервать беременность до 22 недель.
– Бывают случаи, когда генетические анализы не показывают никаких отклонений, а у ребенка все равно появляются недуги, связанные с хромосомными аномалиями?
– Есть отдельная группа хромосомных аномалий – микроделеционные синдромы, которые могут себя «не проявлять» во время беременности. На пренатальном скрининге в группу высокого риска женщина не попадет ни по биохимическим показателям, ни по эхографическим маркерам. Это происходит потому, что либо микроделеционный синдром не сопровождается врожденными пороками развития, либо порок развития имеет позднюю манифестацию, обнаружить его на ультразвуке можно после 22 недели беременности. Чаще всего микроделеционные синдромы, то есть потеря небольшого участка хромосомы, возникает de novo – впервые.
Фото: pixabay.com
– Недавно мировые СМИ рассказывали о мальчике, у которого из-за редкой генетической аномалии почка опустилась в бедро. С какими редкими отклонениями Вы встречались в своей практике?
– У мальчика обнаружена хромосомная аномалия – потеря участка короткого плеча хромосомы 7 – делеция, которая в большинстве случаев возникает de novo – впервые. Каким образом будет себя «проявлять» делеция, зависит от размера потерянного участка и того, какие гены утрачены. «Уникальность» данного случая в том, что ранее у детей с таким же синдромом не выявляли эктопического расположения почки за пределами брюшной полости.
В своей практике я встречалась с разными орфанными заболеваниями в силу того, что консультировала детей, а 50% врожденной и наследственной патологии выявляется в детском возрасте. Это и синдром Вольфа-Хиршхорна, и синдром Моута-Вильсона, и синдром Вильямса, и муколипидоз 2 типа, а также множество других наследственных заболеваний.
Все случаи разнообразные по клинической картине.
– Наука развивается стремительно: говорят, скоро врачи научатся «редактировать» гены у эмбриона. Неужели такое возможно?
– На данном этапе проводится исследовательская работа по редактированию генома. Не исключено, что в ближайшем будущем это будет осуществимо на эмбрионах.
– В настоящее время можно как-то повлиять на генетику будущего ребенка? Например, снизить вероятность рождения малыша с синдромом Дауна у женщин после 40 лет.
– Нет, снизить вероятность рождения ребенка с синдромом Дауна нельзя. Простая трисомная форма возникает в результате случайной «ошибки» – нерасхождения хромосом – во время деления ядер половых зародышевых клеток. В норме из одной зародышевой половой клетки образуется 2 клетки, каждая несет 23 хромосомы. При оплодотворении нормальным спермием, также несущим 23 хромосомы, образуется эмбрион с нормальным численным набором – 46 хромосомами.
Если же при делении происходит ошибка, тогда в одну половую клетку попадает 3 хромосомы, то есть одна лишняя, а в другую – ни одной. При оплодотворении яйцеклетки с тремя хромосомами образуется трисомия, и ребенок будет болен.
Нерасхождение хромосом в мейозе – это случайное событие, предугадать его невозможно. Примерно в 90% случаев ошибки при делении происходят во время оогенеза – деления женских половых клеток, и в 10% во время сперматогенеза – деления мужских половых клеток.
– Какой процент среди причин бесплодия приходится на генетические отклонения?
– Около 50-70% случаев ранней остановки развития и внутриутробной гибели эмбриона обусловлено хромосомными аномалиями, такими как аутосомные трисомии, полиплоидия, моносомия Х-хромосомы и несбалансированные структурные перестройки. В семьях с бесплодием и потерями беременности на ранних сроках частота носительства сбалансированных хромосомных аберраций – 8%.
– Какова осведомленность россиян в вопросах генетики? Они понимают, что целесообразно тратить деньги на генетические анализы, которые все-таки недешевые?
– Осведомленность низкая и по сей день, однако за последние годы благодаря работе генетиков, благотворительных фондов, СМИ ситуация улучшается. Все больше врачей смежных специальностей направляют на консультацию к генетику по разным вопросам.
– Анастасия Владимировна, почему Вы выбрали такую специализацию, как репродуктивная генетика? Мне, как неспециалисту, кажется, это очень сложно, но в то же время интересно.
– На самом деле я клинический генетик, а значит, области моего консультирования разные. Я всегда стремилась работать не только со взрослым населением по вопросам репродукции и пренатального скрининга, но и активно консультировала детей. И даже некоторое время работала в стационаре – в отделении врожденных и наследственных заболеваний. Эта внутрисистемная работа в области медицинской генетики крайне важна для развития специалиста.
Источник
В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний — бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) — врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента — меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.

Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.
Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя — третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией — трисомией по 18 паре хромосом. Причина — нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
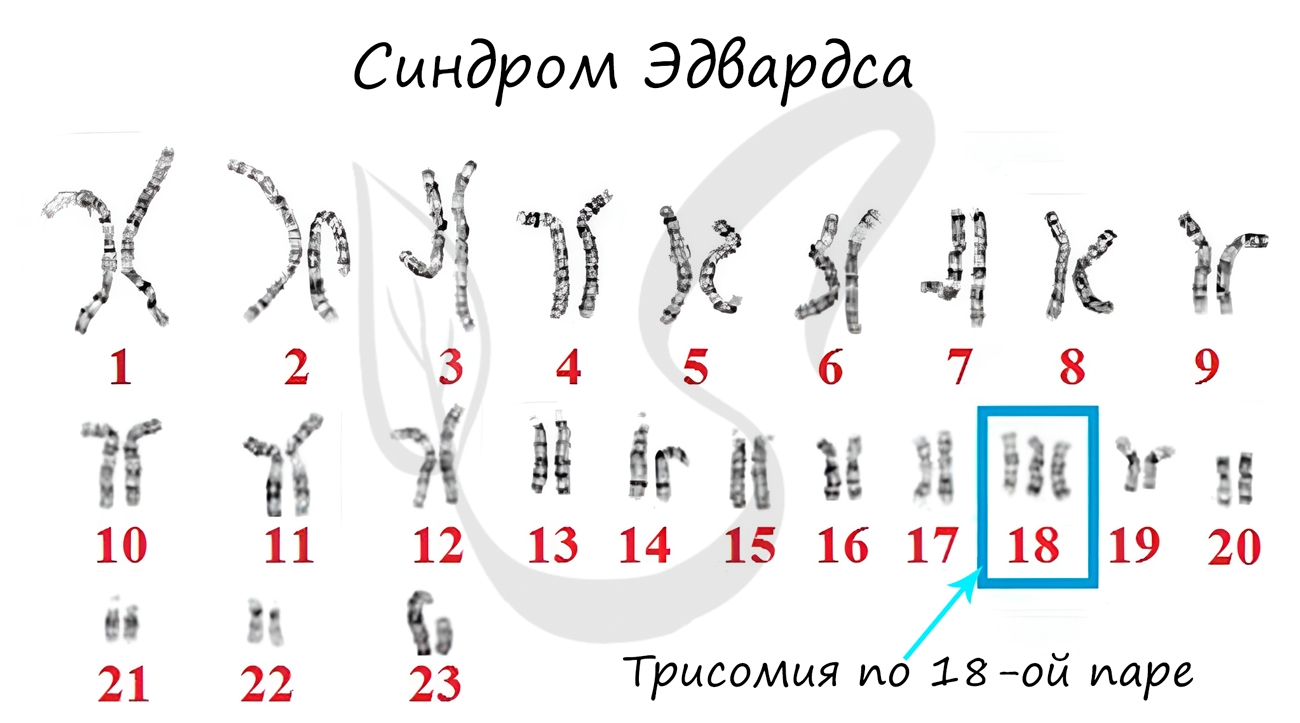
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией — трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
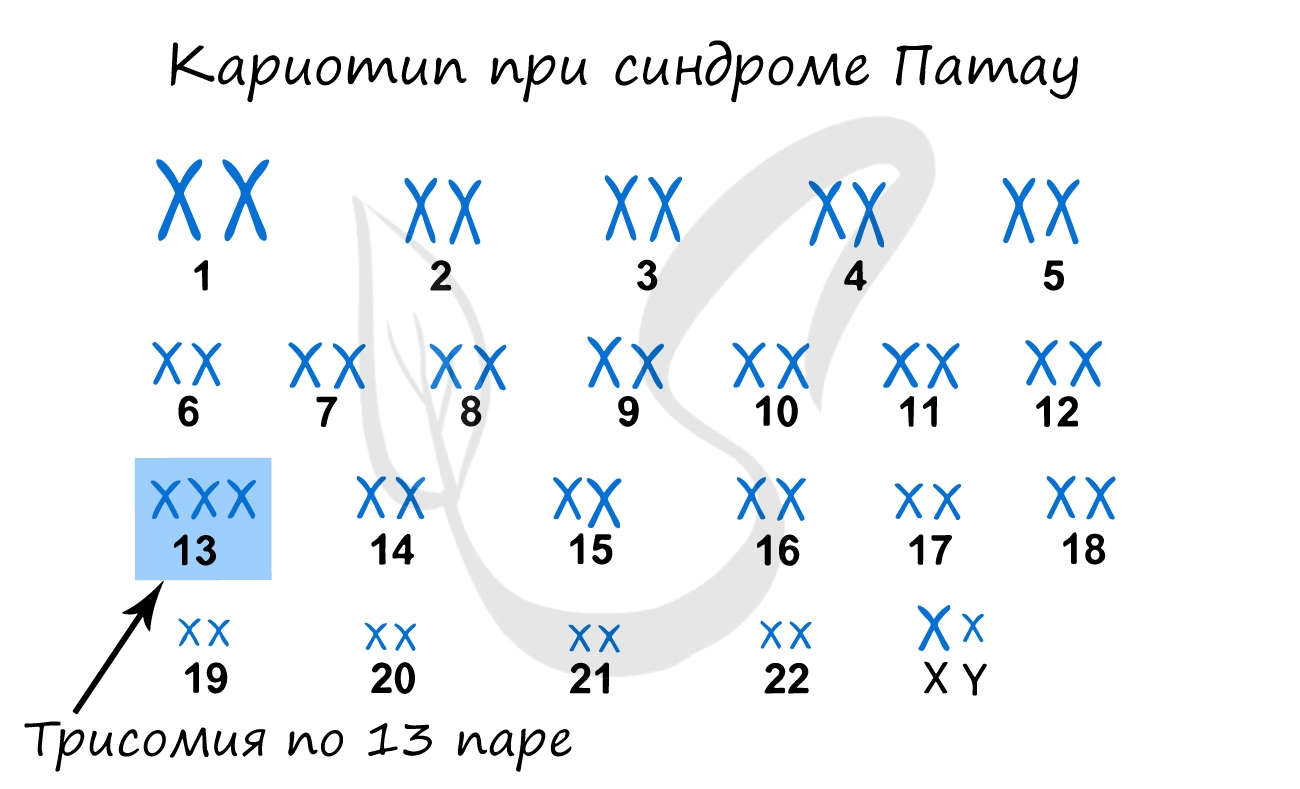
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа — «XY» 🙂
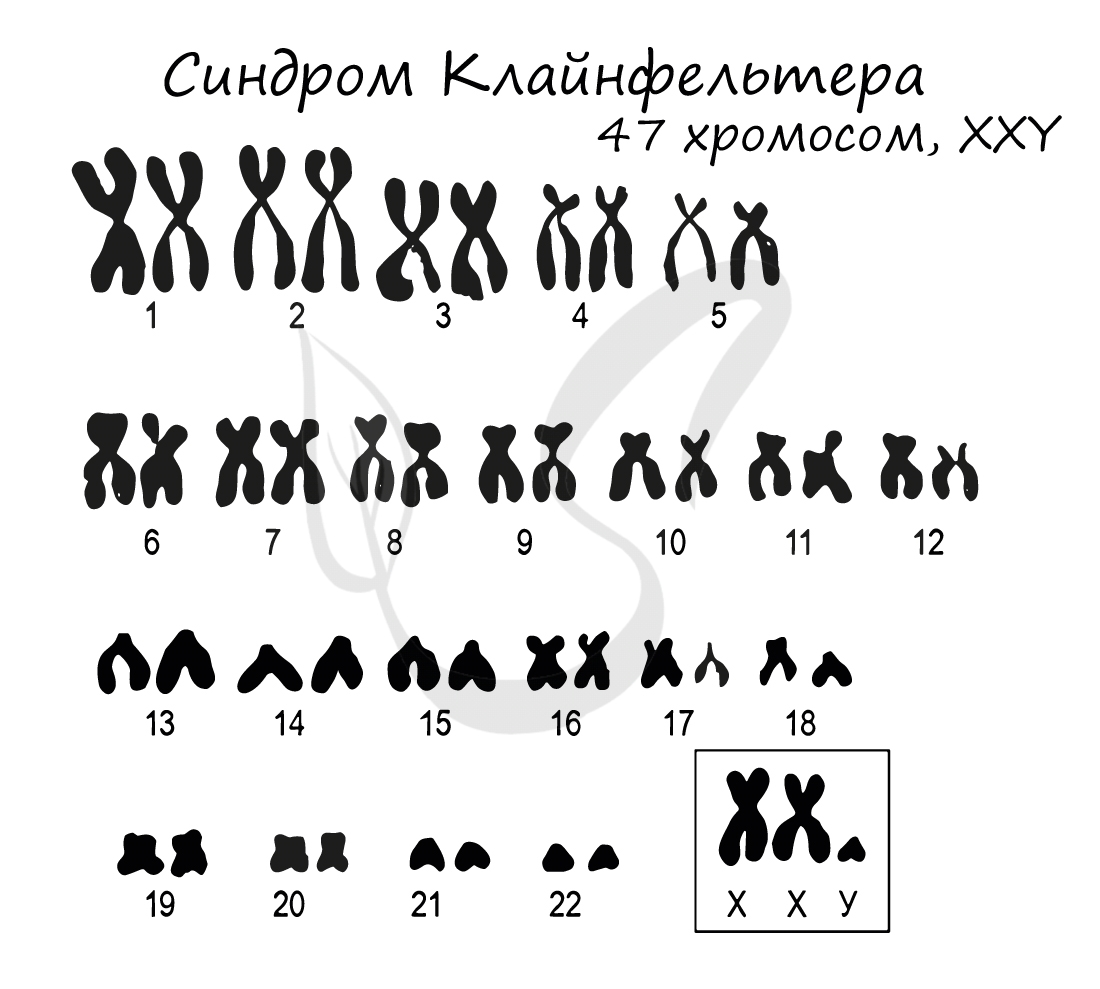
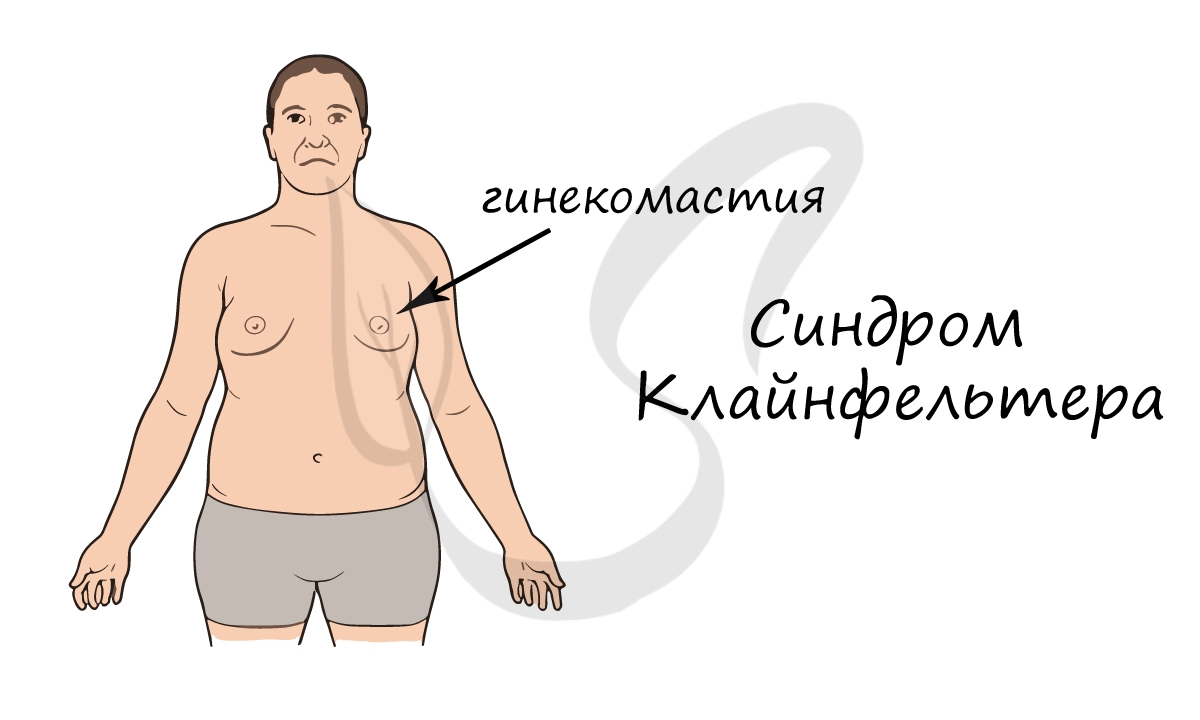
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера — наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения — эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2020
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник